Annotating with gnomAD: Frequencies from 123,136 Exomes and 15,496 Genomes
When the Broad Institute team lead by Dan MacArthur announced at ASHG 2016 that the successor to the popular ExAC project (frequencies of 61,486 exomes) was live at http://gnomad.broadinstitute.org/, I thought their servers would have a melt-down as everyone immediately jumped on and started looking up their favorite genes and variants.
But although the web interface was up, it would be a long four months until the downloadable bulk variant frequencies were made available.
We have just recently finished our curation and QC of these tracks, and have made them available today for usage in VarSeq, SVS and GenomeBrowse.
Bigger and Better
At first I didn’t get why gnomAD needed a new website and a new brand for what was essentially “ExAC 2.0”. But having spent some time with the data, I understand the need for a clean break.
ExAC is BACK! @dgmacarthur now discussing round 2 of ExAC: we double the sample size and add WGS (126,216 exomes + 15,136 genomes) #ASHG16
— Konrad Karczewski (@konradjk) October 19, 2016
This really is a new “product” from a data processing perspective, and given the cohort itself has been expanded to a wider selection of ethnicities and the data processing pipeline updated to include new and novel ways of flagging low-quality variants, a new name is very much appropriate.
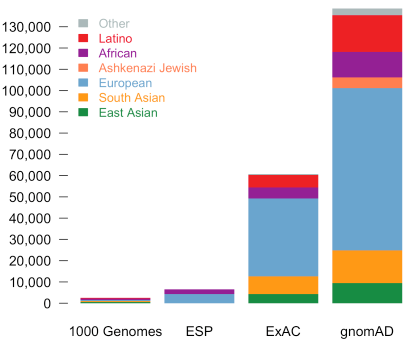
Compared to ExAC, there is the entirely new population category of Ashkenazi Jewish and greatly expanded numbers in all population sub-groups.
If you want to understand more about the project and the analytics that produced the variant calls and the QC flags around them, check out the release announcement blog.
Data Curation: Making Annotation Precise and Understandable
There are several improvements we added over the raw VCF file as we curate this track for the explicit purpose of supporting the variant interpretation done by our clinical and research users.
- Split multi-allelic sites into single-alternate allele records to allow precise annotation
- Map the correct QC flags and per-allele values to these individual records
- Document and organize the fields and the source
- Left-align using Smith-Waterman to ensure every representation of a variant can be annotated
The first is probably the most important. By providing their variant “site” annotations as a VCF group, the gnomAD team is following the VCF spec that says only a single line can have a given start position and reference.
Of course, there are plenty of sites where the multiple mutations occur independently at the same location at the genome.
For example, these pairs of variants in BRCA2 (one common, one rare):
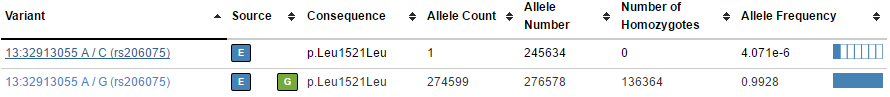
In the VCF file, these two variants are on the same row with per-allele information such as frequency (AF) turned into a list that matches each corresponding ALTA allele.
| CHR | POS | ID | REF | ALT | QUAL | FILTER | INFO |
| 13 | 32913055 | rs206075 | A | G,C | 5.18E+08 | PASS | AC=244277,1;AF=9.94476e-01,4.07110e-06;AN=245634;… |
When annotating this variant, we would like to annotate our single variant in our sample (say a G/C) with the more precise single records as they appear on the web interface.
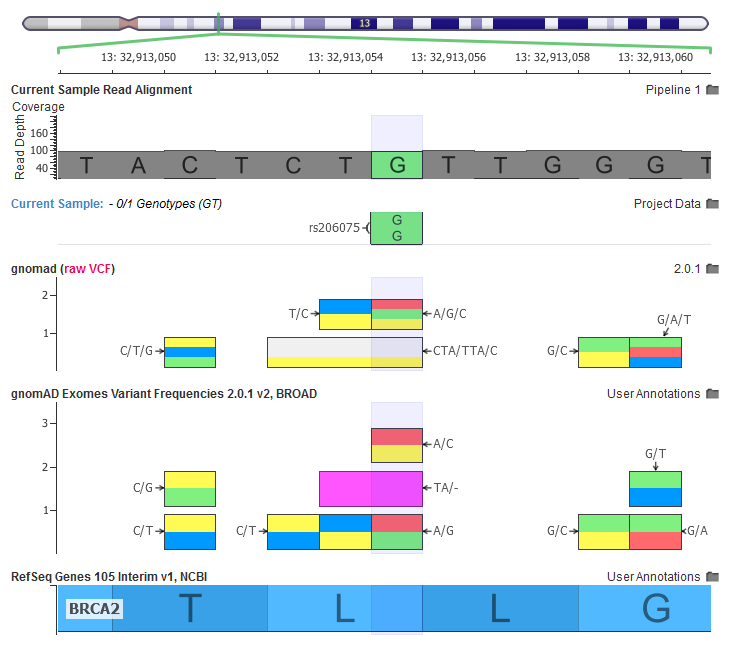 The current sample has a homozygous variant, the gnomAD RAW VCF file has a multi-allelic variant, our curated gnomAD annotations breaks out each multi-allelic site into their own records.
The current sample has a homozygous variant, the gnomAD RAW VCF file has a multi-allelic variant, our curated gnomAD annotations breaks out each multi-allelic site into their own records.
This allows precise annotation of the allele specific information from gnomAD for the A/G variant.
Not only does this include common things like Allele Frequency, but also other per-allele specific fields including Allele Counts, Homozygous genotype counts (by sub-population) and QC metrics such as Read Depth of allele carriers (discussed below).
In the above screenshot, we can see how multiple multi-allelic variants are being transformed through our curation scripts. Along with the A/G/C variant being broken out into a A/C and A/G, we also see a CTA/TTA/C variant being broken out to a C/T and a TA/- (two letter deletion).
An Eye on Quality
In a recent post about the Filter fields in ExAC, I made a case that is important to pay attention to the quality flags computed on these population catalogs.
In gnomAD, these field remain critically important and have been expended with some new flags:
- RF: This is the output of a machine learning (random forest) algorithm that predicts which variants are likely to be false-positive calls
- LCR (Regional): These variants fall within Low Complexity Regions of the genome, often composed of simple repeat sequences
Finally, there are some other summary fields that provide supplementary details to gauge the quality of the variant gnomAD is calling:
| Alt Allele Prob (RF) | 0.176659 |
| Alt Carrier DP Median | 21 |
| Alt Carrier GQ Median | 67 |
| Het Carrier Allele Balance Median | 0.397917 |
| Ref Carrier Dosage Median | 0.000125563 |
This Variant was flagged as PASS, but has other metrics that might make you question its validity.
These are useful enough to go into more detail:
- The “Alt Allele Prob” is the probability based on the Random Forest classifier that the variant is high quality. It will be close to 1 for high-quality variants and close to 0 for low-quality variants.
- The first two median scores are the median Read Depth (DP) and Genotype Quality (GQ) of the samples that have the variant.
- The Allele Balance Median score is the ratio of the reference and alternate allele in the reads for samples that have the variant called as Heterozygous (which you expect to be very close to 0.5).
- The Dosage Median (frequency of the alternate allele) for samples that were called reference, which you would expect to be very close to 0.
Stay Tuned
We found the Low Complexity Region flag so useful in our analysis that we will be curating it as a separate track you can use to annotate all of your variants.
Although the gnomAD team is working on starting from scratch and re-aligning and calling using the GRCh38 reference, we think it would be useful to provide an intermediate solution for folks using the new human reference genome. To that end we are working on a LiftOver like algorithm that maps variants to GRCh38, as well as corrects for reference allele changes that will affect the variant annotation in the process.
Stay tuned for further announcements on that topic!



Great blog and post.
The two points that seem to finally be getting real attention by the community (but still not quite there) are
the utility of
“Splitting multi-allelic sites into single-alternate allele records to allow precise annotation”
and
“…a LiftOver like algorithm that maps variants to GRCh38, as well as corrects for reference allele changes that will affect the variant annotation in the process”
The ClinGen grant in conjunction with NCBI’s ClinVar have been addressing this issue for several years. IMO it is a fundamental concept that will help open up the field for sharing data (not just annotations for research) but for actual clinical care by providing EHRs with the capability to utilize well specified variant findings to provide providers with the best available and up to date “expertly” curated knowledge from multiple “clinically-approved” resources.
We go as far as suggesting that the “LiftOver” algorithm can be a stepping stone to developing an equivalency specification that would enable the development of canonicalized alleles which will auto-map to all the well-known and available reference assemblies going forward. ClinGen has an Allele Registry developed just for this purpose and we are continuing to utilitize it to demonstrate the potential power and benefits not just for curation/annotation, but ultimately for enabling EHR vendors to have a fighting chance of exposing the the utility of genetic test results.
see http://www.clinicalgenome.org for info on ClinGen
see http://reg.genome.network/ for info on the ClinGen Baylor Allele Registry
see datamodel.clincalgenome.org for info on the modeling work behind these allele and interpretation models.
Thanks for communicating the extremely helpful points to the community!