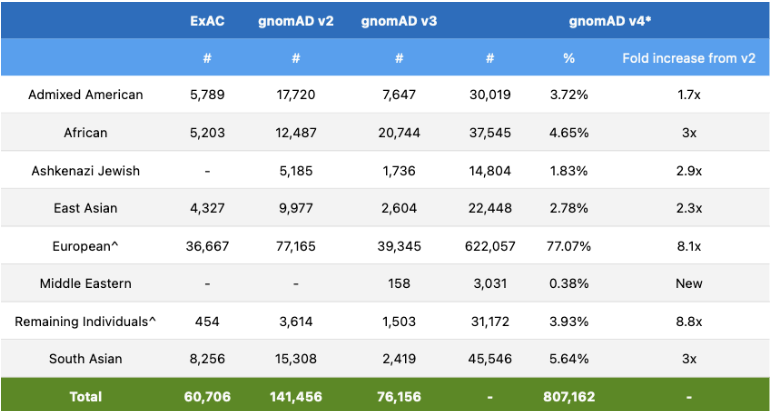
The Broad Institute’s release of gnomAD v4 needs no introduction as the data in this release is highly sought after by professionals in the genetics community, and the v4 release has a lot to boast about! The v4 release is roughly five times larger than the v2 and v3 releases combined and includes data from 807,162 total individuals. Naturally, exome sequencing data of this size introduces global diversity and includes about 138,000 individuals of non-European genetic ancestry. However, this release also included 416,555 individuals from the UK Biobank, which increases the proportion of samples with European ancestry compared to previous releases.
A new data feature included in the gnomAD v4 release annotates the variants within the database with clinical and functional effects from REVEL, CADD, PhyloP, spliceAI, and Pangolin. For more details on curation updates that The Broad Institute included in gnomad v4, check out this blog post!
Golden Helix was just as excited as all of you were when the gnomAD v4 data was released in November. Eagerly, we began data curation. So why are we just now introducing the v4 release to our Golden Helix customers? GnomAD is a wonderfully large aggregation of data, broken up into per-chromosome files with many more fields than can be used for a single use case. The raw genome data files alone weigh in at roughly 830GB! After the files are downloaded, the real work begins. Our curation team invested a large amount of time in generating a script that cleans up the raw data files so that the gnomAD annotation source is intuitive to our VarSeq users and compatible with VarSeq and VSClinical. Our curation script includes a number of data refinement steps ranging from generating meaningful field names to filtering out flagged variants that do not have allele counts (AC0 Variants) to minimize the size of the annotation file.
For the gnomAD v4 release, we did a little bit more curation work than usual to make sure our Golden Helix customers have high-quality annotation sources and can maximize gnomAD’s data in their VarSeq workflows.
- Creation of the GRCh37 gnomAD v4 tracks. The gnomAD v4 released data is native to the GRCh38 reference genome. A great deal of effort was put into improving our LiftOver algorithm so we could generate gnomAD frequencies for both GRCh37 and 38 users. Recently, we published a blog discussing the details of the gnomAD v4 liftover to GRCh37, including how it is necessary to handle the cases where the reference allele changes between genome versions. Of course, no lifted-over track is as perfect as one generated from natively aligned data, but we believe the Golden Helix gnomAD v4 GRCh37 track is the best possible representation of the gnomAD data on the 37 reference and a good default for annotation and filtering workflows.
- The new gnomAD v4 “joint” track. In the past, we have had customers request a version of the gnomAD database that merges the exome frequencies and genome frequencies into a single harmonized source. This merging would aggregate allele counts when a variant is present in both the exomes or genomes and pass through variants unique to either source. In gnomAD v4, the team at the Broad computed these merged frequencies themselves and added an extra set of “joint” fields to both the raw exome and genomes’ VCF files. For this release, we curated a “Joint Variant Frequencies” track based on these fields and merged without duplication all the variants from the disparate exome and genome raw sources to create a single comprehensive source.
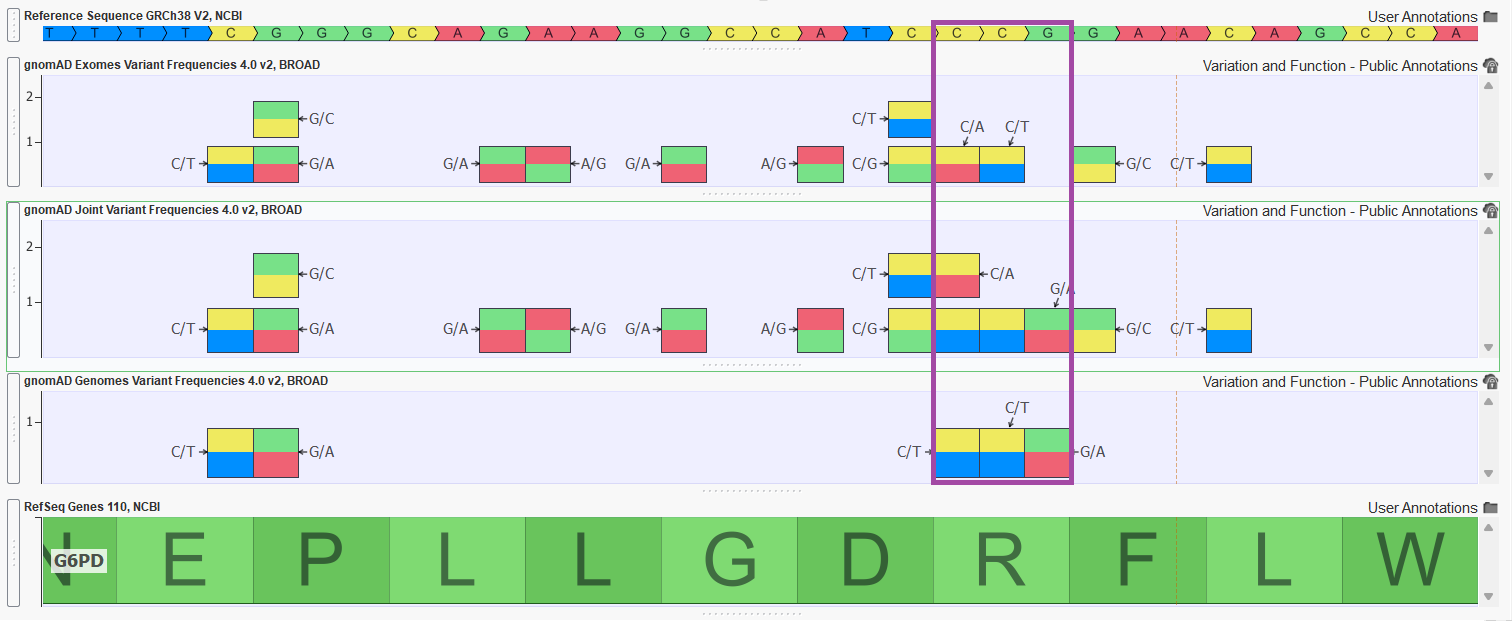
Figure 2 shows a handful of variants in exon 4 of the G6PD gene. Notice how the joint track is capturing variants that are unique to gnomAD exomes and gnomAD genomes but also merging variant entries to a single entry in the joint track (merging allele counts and frequency calculations as well).
The gnomAD Joint Variant Frequency tracks are so much more comprehensive than each of the Exomes, and Genomes tracks individually that in the upcoming VarSeq 2.6.0 release, we have incorporated the gnomAD Joint Frequencies track into the ACMG and Cancer Classifier algorithms and into all shipped VarSeq project templates that include a gnomAD frequency track.
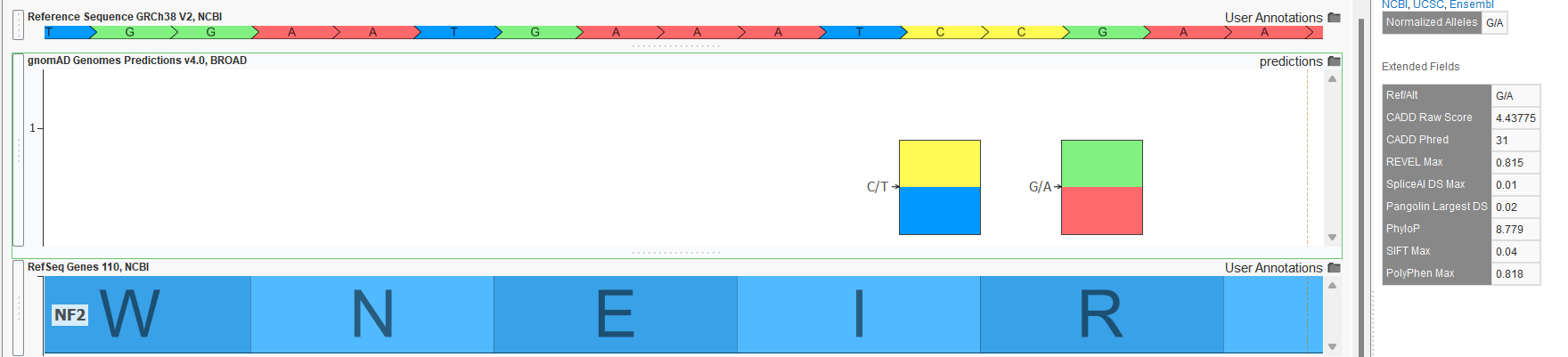
- Functional and splice predictions in the gnomAD Joint Predictions track. The raw VCF files for the exome and genome sources also contained various functional predictions computed by the Broad. The computed scores include REVEL, CADD, PhyloP, SIFT, PolyPhen, spliceAI, and Pangolin. A Joint Predictions track was created with only these prediction scores present for all variants in either the exome or genome raw VCF file. No “AC0” filtering was done, so variants may be present in this track that are not called in the variant frequency tracks. Figure 3 shows the output for the G/A variant in the NF2 gene in GenomeBrowse. Of course, if a project is annotated with this source, these fields could be included in variant filtering logic with scoring thresholds.
We are very excited for our Golden Helix customers to be able to access and use the gnomAD v4 annotation sources in their VarSeq workflows. A lot of hard work has gone into curating the best gnomAD annotation thus far from both The Broad Institute and the Golden Helix curation team. Still to come are the structural variant, and gene constraint gnomAD v4 sources, which are currently in the process of being curated. If you have any questions about the gnomAD resources that are available or adding gnomAD to your VarSeq projects, please reach out to us at [email protected].



