Sentieon secondary analysis, inside VarSeq
Sentieon runs alignment, deduplication, and variant calling at 10X-50X the throughput of open-source BWA-GATK, with deterministic, mathematically equivalent results. DNAscope adds pre-trained ML models for short-read and long-read calling, and hands the VCF directly to VarSeq for tertiary analysis.
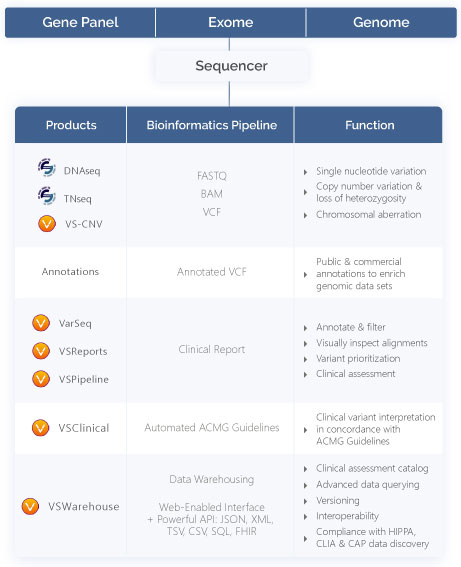
Secondary analysis NGS turns raw sequencer output into a structured variant table. The input is millions of reads in FASTQ format; the output is a list of variants relative to a reference genome, in VCF.
Achieving high-throughput in clinical genomics requires variant calling software that balances speed, accuracy, and reproducibility. Legacy pipelines often face bottlenecks in processing whole-genome and whole-exome datasets, leading to extended turnaround times and hardware scaling challenges.
Golden Helix integrates Sentieon as the DNA sequencing pipeline engine in the VarSeq Suite for alignment, deduplication, and variant calling. DNAseq and TNseq match GATK and MuTect2 mathematics. DNAscope adds pre-trained ML models that improve sensitivity through better active-region detection and local assembly, covering both short-read and long-read sequencers.
What Is High-Performance Variant Calling?
Modern genomic research demands a high-performance variant calling approach that can scale to national-level genome projects. Sentieon achieves this through optimized software implementation of the GATK Best Practices workflows.
- BWA Alignment: Reads map to the reference with BWA-MEM or BWA-MEM2, optimized for multi-core throughput on standard CPUs.
- Deduplication: Identifying PCR artifacts and marking redundant copies to improve variant calling accuracy and reduce data volume.
- Variant Calling: Statistical models examine read pileups to determine genotypes using Bayesian inference for SNVs and Indels. DNAscope incorporates pre-trained machine learning models for improved accuracy.
Why Sentieon Outperforms Legacy Tools
Two specific properties matter for high-throughput labs and clinical validation: results match GATK, and they match themselves across reruns.
- Mathematical Equivalence: Implements the exact same mathematics as BWA-GATK and MuTect2, achieving mathematically equivalent results with massive throughput gains.
- DNAscope ML Caller: Pre-trained machine learning models provide improved accuracy through enhanced active region detection and more powerful local assembly of reads for both short-read and long-read sequencers.
- 100% Deterministic: The same input always produces the same output, which lets clinical validation lock a pipeline version and reproduce it on demand.
- Generic CPU Support: Pure software solution that runs on standard CPUs, with no specialized GPUs or expensive hardware accelerators required.
What to Look for in Secondary Analysis Software
Throughput
10X-50X fewer core-hours than standard Java BWA-GATK on the same FASTQs, so a clinical exome backlog clears in a shift instead of a day.
Reproducibility
Deterministic results, with no downsampling in high-coverage regions. Validation runs and production runs agree call-for-call.
Population Scale
Joint calling on 100,000+ samples without staging intermediate merge files. The same binary runs for a single trio or a national cohort.
Germline and Somatic
DNAseq for germline and TNseq for tumor-normal somatic calling, in one engine and one license.
FASTQ to Report
Sentieon hands the VCF directly to VarSeq in the same install, so secondary and tertiary share one project and one audit trail.
Deployment
Run on a workstation, a server, or an air-gapped network. Pure software, standard CPUs, no GPU dependency.
Sentieon and VarSeq, one install
Golden Helix and Sentieon have run a multi-year integration so labs get Sentieon's runtimes without changing their scientific output. Sentieon turns the sequencer's FASTQs into a VCF; VarSeq picks that VCF up in the same project for filtering, annotation, and reporting.
10X-50X Faster Pipeline
10X faster FASTQ-to-VCF and up to 50X faster BAM-to-VCF on the same hardware as a standard BWA-GATK pipeline.
Clinical Grade Accuracy
DNAseq and TNseq match GATK and MuTect2 call-for-call. DNAscope's ML caller raises sensitivity and specificity on both short-read and long-read data.
VSPipeline Automation
VSPipeline drives Sentieon and VarSeq end-to-end, so a finished run goes from sequencer to clinician-ready PDF without an analyst in the loop.
Sentieon hands an analysis-ready VCF to VarSeq inside the same project, so tertiary analysis starts the moment secondary finishes.
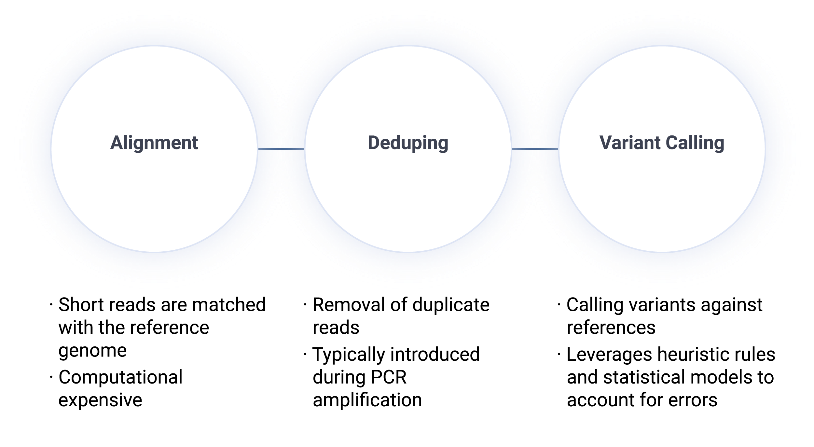
The Sentieon Secondary Analysis Workflow
Read Alignment
Map sequencing reads to the reference genome using BWA-MEM or BWA-MEM2.
Deduplication
Identify and mark PCR artifacts to ensure variant calls are based on unique biological fragments.
Variant Calling
Apply Bayesian models (DNAseq) or ML-optimized calling (DNAscope) to identify SNVs and Indels with deterministic, 100% consistent results.
VCF Generation
Produce standard VCF files ready for clinical-grade tertiary analysis and interpretation in VarSeq.
Enterprise Scaling for Population Genomics
Sentieon is designed for massive datasets, supporting joint calling on 100,000+ samples without intermediate file merging.
- Pure Software Solution: Runs on standard CPUs, with no specialized GPUs needed.
- No Downsampling: Uses every read even in ultra-high coverage regions.
- WGS & WES Ready: Optimized for whole-genome and whole-exome scale data.
Germline & Somatic Solutions
Germline DNAseq & DNAscope
Complete solution for germline SNV and Indel detection. DNAseq provides mathematically equivalent GATK results at 10X speed, while DNAscope uses ML models for even higher accuracy on both short and long reads.
Variant InterpretationSomatic TNseq
Tumor-normal somatic calling matching MuTect and MuTect2 mathematics, at the same throughput Sentieon delivers on germline runs.
Somatic SolutionsPopulation Joint Calling
Scale to national genome projects with joint calling for 100,000+ samples. Deterministic results ensure data consistency across cohorts.
Whole Genome AnalysisFrequently Asked Questions
Are Sentieon results compatible with GATK?
Yes. Sentieon's DNAseq implements the exact same mathematical models as Broad Institute's Best Practice Workflows, achieving mathematically equivalent results with massive throughput gains. DNAscope goes beyond GATK equivalence by incorporating pre-trained machine learning models for improved accuracy on both short-read and long-read data.
Does Sentieon require GPUs for acceleration?
No. Sentieon is a pure software solution optimized for standard CPU architectures. It delivers 10X-50X performance gains without requiring specialized hardware like GPUs or FPGAs.
What is meant by "100% deterministic results"?
Unlike some bioinformatics tools that may produce slightly different results across multiple runs due to multi-threading randomness, Sentieon ensures that the exact same input always produces the exact same output.
Can I automate Sentieon within my existing pipeline?
Yes. Sentieon is highly modular and can be integrated into VSPipeline or custom automation scripts, enabling a hands-off workflow from FASTQ generation to clinical reporting.
Secondary Analysis Insights & Webcasts
Learn how to optimize your NGS pipeline for speed and accuracy with Sentieon and VarSeq.
See Sentieon run on your FASTQs
Talk to our team about deploying Sentieon inside VarSeq, with the same throughput numbers other high-throughput labs and national genome centers run today.