We are happy to announce that our latest version of SVS includes the ability to call CNVs on low read depth Whole Genome Sequencing (WGS) data. Designed for calling large cytogenetic events, this algorithm can detect chromosomal aneuploidy events and other large events spanning one or more bands of a chromosome from genomes with average coverage as low as 0.05x. In this blog post, we will be discussing considerations to take into account when calling CNVs from shallow WGS data in SVS and VarSeq, as part of a broader CNV analysis workflow.
Bin Size
The bin size is an important consideration when calling CNVs from WGS data that can greatly impact the quality of your results. The WGS CNV caller in VarSeq was designed specifically to call large events spanning one or more bands of a chromosome from low coverage whole genome data. As a result, the algorithm’s accuracy is closely tied to the selected bin size. Larger bins allow the noise within each bin to be smoothed over, thereby lessening the effect of coverage fluctuations. Additionally, in the case of shallow whole genomes, large bin sizes ensure that each bin contains a sufficient number of reads, allowing large-scale fluctuations in coverage to be reliably identified. In contrast, smaller bin sizes can often result in more false positives and erroneous segment boundaries for higher coverage genomes and lost signal causing large events being missed entirely for low coverage genomes.
Shallow whole genome data is especially susceptible issues arising due to small bin sizes. In the images below, we show two sets of Z-score values for a low coverage whole genome sample with an average read depth of 0.05x. This sample contains a chromosome 22 aneuploidy event. In the first image, we used a bin size of 1 million base pairs and the signal indicating the duplication event is extremely clear.
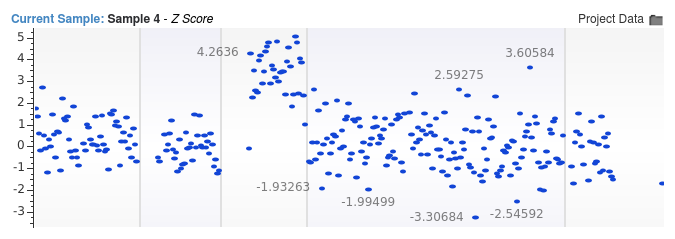
In the second image, we used a bin size of only 10,000 base pairs.
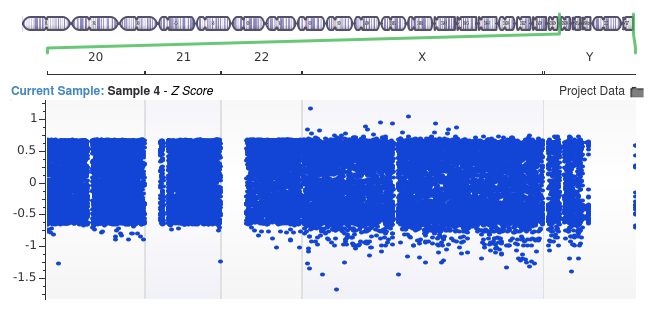
Notice that the signal indicating the duplication event has vanished entirely. Because the average coverage for each bin is either zero or extremely close to zero, all of the regions appear to differ very little from one another. As a result, the Z-score values never indicate coverage fluctuations surpassing more than a single standard deviation from the mean. Thus, it becomes impossible to call the event.
In addition to impacting the quality of called events, a smaller bin size will also increase the required runtime of the algorithm, as the algorithm’s computational performance is directly tied to the total number of bins. Generally, for shallow whole genomes, we recommend a bin size of around 1,000,000 base pairs.
Segmentation Algorithms
A relatively recent feature for our whole genome CNV caller is the ability to select between two different segmentation methods. The first is an approximation algorithm called Circular Binary Segmentation (CBS) which performs segmentation by iteratively computing segments to maximize the variance between segments while minimizing the variance within each segment. The second is our internally developed CNAM optimal segmentation algorithm, which exhaustively searches over all possible cut-points for a given region of data, finding the set of cut-points that minimize the sum of squared deviations of the data from the mean of its respective segment. While CNAM is guaranteed to produce a provably optimal segmentation of the data, CBS is generally preferred, as it typically produces comparable results in a fraction of the time required by optimal segmentation.
Annotation and Filtering
When calling CNVs from WGS data in VarSeq, you get access to the complete annotation and filtering functionally that our customers use across genomic analysis workflows, including filtering small variants and CNVs called from gene panel and exome data. This includes support for a wide variety of variant annotation tracks, including 1kG CNVs and Large Variants, which provides frequency information about CNVs, ClinVar, which provide clinical interpretation information about CNVs, Genomic Super Dups, which catalogs regions that are highly homologous and problematic for mappability, and gene tracks such as RefSeq Genes and Ensembl. All of these annotations can be filtered on, allowing users to reduce false positives and filter out common CNVs.
Conclusion
Hopefully, this blog post has provided some useful information about our CNV caller for WGS data. If you are interested in trying out our CNV caller or are interested in adding CNV to your existing VarSeq or SVS licenses, please reach out to [email protected]. Our team of experts would be happy to demonstrate how you can use this powerful feature.



