
Revolutionize Your Somatic Variant Analysis with Our Cutting-Edge Template for Annotation and Filtering in VarSeq
Golden Helix is excited to share our new Comprehensive Cancer Template for somatic variant annotation and filtering, along with the latest version of our software VarSeq 2.3.0! Our latest VarSeq update was specifically focused on getting up to speed with multiple aspects of somatic variant analysis and clinical cancer genomics. Thus, it is only fitting that we released an up-to-date, practice-wide customizable template that users can leverage as a practical starting point for their somatic variant annotation and filtering.
The Comprehensive Cancer Template
The Comprehensive Cancer Template was designed for importing and filtering large cancer gene panels in preparation for evaluation in VSClinical AMP, especially for TSO-500 and other comprehensive genomic profiling tests. The filtering process was designed based on the strategy laid out in a 2019 article1 by the ComPerMed working group, a panel of Belgian experts in cancer diagnostics that set out to establish a uniform biological classification system and streamline clinical interpretation of somatic variants detected by NGS. The VarSeq filtering strategy is mainly centered around whether a variant falls in a tumor suppressor gene or an oncogene. Several ComPerMed recommendations are addressed in the Comprehensive Cancer Template discussed below. We also address other factors within VSClinical evaluation, such as our clinical reporting formats and Oncogenicity scoring recommendations which cover presence in Somatic Catalogs, in silico predictions, and previous reports of oncogenicity or pathogenicity in Civic or ClinVar. The filter template will do a lot of the heavy lifting of prioritizing variants before import into VSClinical for final oncogenic classification and Biomarker, Drugs, and Clinical Trials evaluation according to the AMP guidelines.
Variant Quality
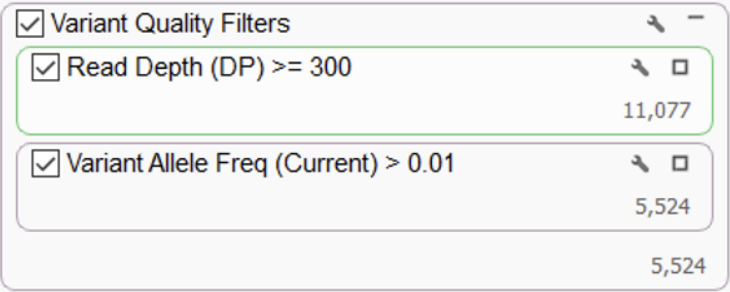
After variant calling, it is highly recommended to apply a technical filter to select variants of high quality and remove any artifacts. Two very fundamental quality filters are used to eliminate potential false positives – read depth and variant allele frequency. These suggested thresholds shown below (Figure 1) are a good starting point. Users are free to set their desired quality thresholds and add any additional quality parameters defined in their lab protocols, especially for the removal of technically generated artifacts.
Variant Function and Filtering
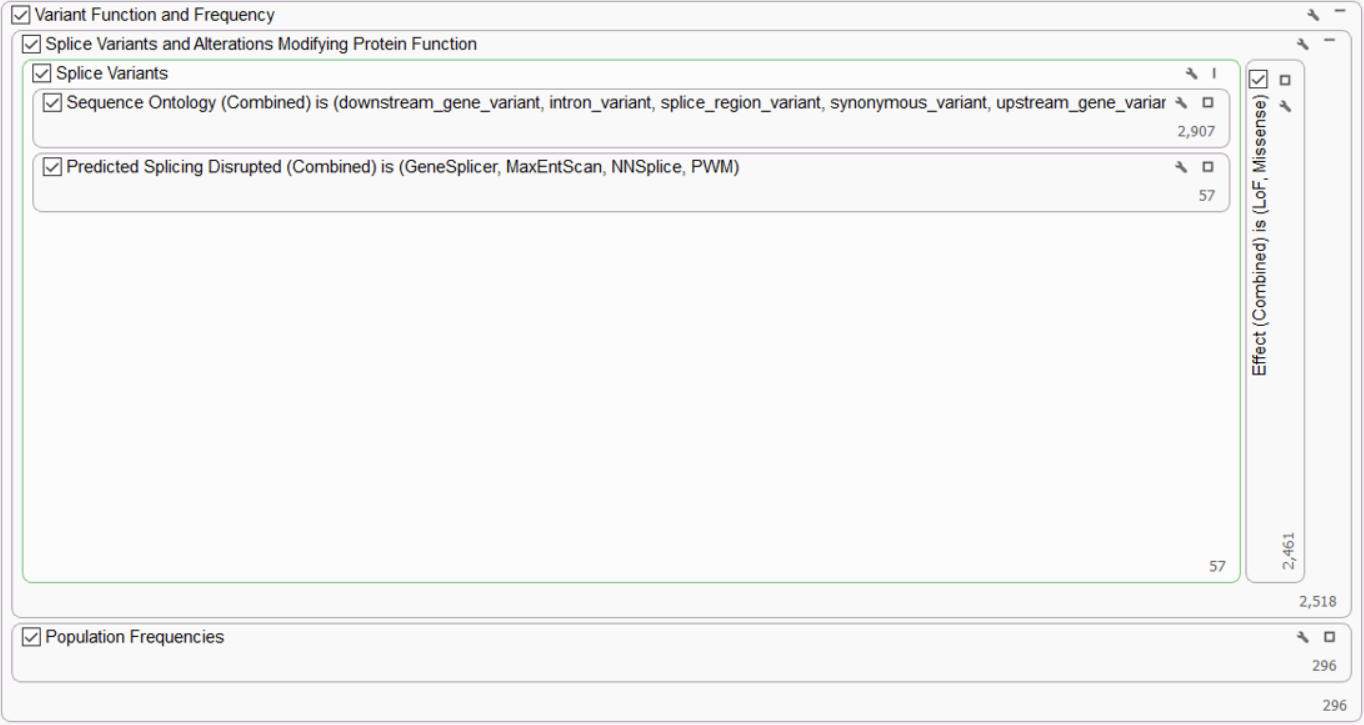
The ComPerMed working group also recommends the removal of variants that are common in the healthy population and the retention of variants that are most likely to affect protein function. Using our template will automatically add recommended annotations such as gnomAD Exomes Variant Frequencies, 1KG Phase 3 Variant Frequencies 5a, and dbSNP Common 155 to the project. We employ these as population frequency filters to scrub out common variants, considering that variants with a minor allele frequency of 0.1% or more are most often benign or likely benign. In this section, we also deploy RefSeq Genes track to identify variants with functional impact on splicing, loss of function, and missense variants.
Somatic Variant Filtering Workflows
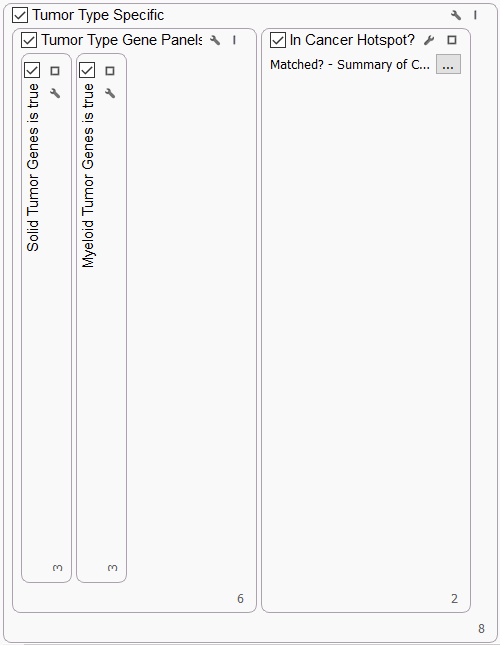
TUMOR TYPE SPECIFIC
The ComPerMed group developed Consensus Pathogenic Variant (CPV) lists of variants in genes required for screening in solid tumors and myeloid tumors. In the Tumor Type Specific section of the workflow, we incorporated the list of genes using our Match Gene List algorithm. This section also captures variants that fall in the Cancer Hotspots list, an interval track maintained by the Kravis Center for Molecular Oncology at Memorial Sloan Kettering Cancer Center.
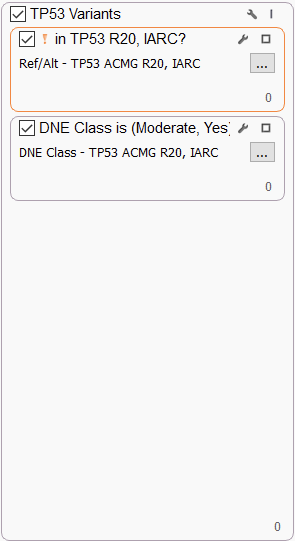
TP53 VARIANTS
The TP53 tumor suppressor gene deserves special consideration because variants affecting almost every position of the protein are detected in multiple tumor types. We use the recommended International Agency for Research in Cancer (IARC) TP53 database in our filter since it contains widespread information on human TP53 cancer variants from peer-reviewed literature and generalist databases. Specifically, we filter on variants in the database that have a definite (“Yes”) Dominant Negative Effect (DNE), i.e., cause dominant-negative activity on both WAF1 and RGC promoters or on all promoters, or a “Moderate” effect, i.e., dominant-negative activity on some but not all promoters.
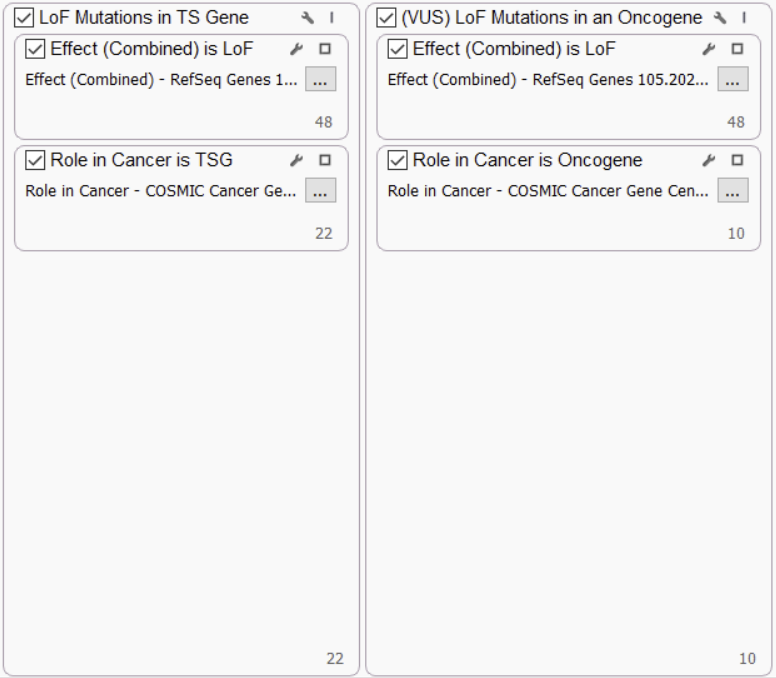
LOF MUTATIONS IN TS GENES AND ONCOGENES
Clear loss of function (LoF) mutations in tumor suppressor (TS) genes are considered likely pathogenic by ComPerMed, while LoF in oncogenes will not likely play a role in cancer, so they are considered variants of unknown significance (VUS). As such, we incorporate these parameters into the filter chain using the Cosmic Cancer Gene Census track to capture oncogenes and TS genes.
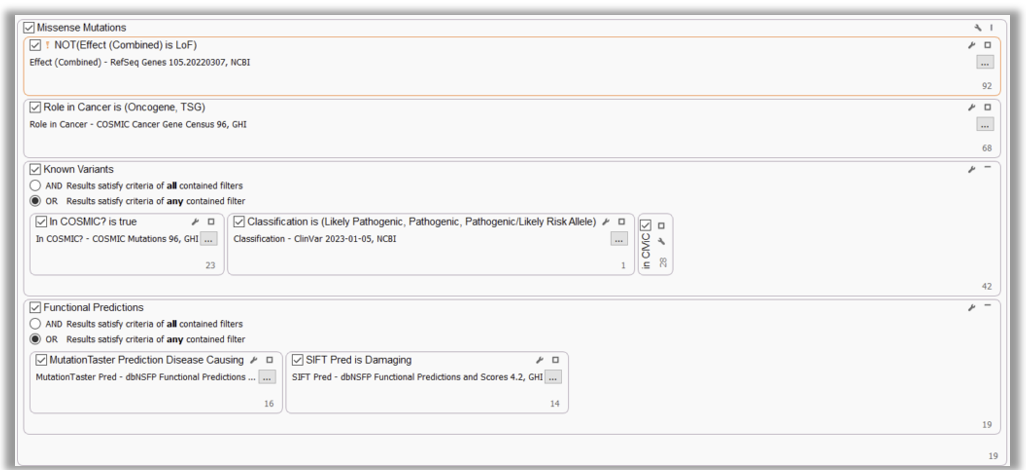
MISSENSE MUTATIONS
It is also highly recommended to consider variants within TS genes and oncogenes that are not clear LoF, for example, missense variants or in-frame indels. These could result in a partial loss OR activation of an allele and potentially be classified as “Likely Oncogenic“ or “VUS. “ Here, we included filters to capture oncogene and TS status and non-LoF gene impact. To make this filter strategy more robust, we’ve also added filters to capture well-known cancer variants (COSMIC) and pathogenic/oncogenic variants (ClinVar/CIViC), and variants that have damaging and disease-causing functional impact predictions (dbNSFP).

This Comprehensive Cancer Template is well-suited for use as a starting point for somatic variant annotation and filtering in the context of both solid and hematological tumors. Users doing NGS in the somatic variant context are free to begin with this template as a framework to create a robust somatic variant analysis workflow. For questions or training regarding the use of this filter template, please reach out to us at [email protected].




{kind=link}