During the webcast yesterday, I demonstrated a few ways of customizing de Novo Candidate and Compound Heterozygous Candidate workflows to consider family structure that was slightly different from the default trio workflow. The families included additional affected and unaffected siblings added to a trio as well as looking at what could be done if there were only two affected siblings and no parents.
Each custom workflow in VarSeq used different existing features and some soon to be released features to filter and prioritize variants. Additionally, we used a customized Clinical Report template to showcase reporting variants from a Quad analysis.
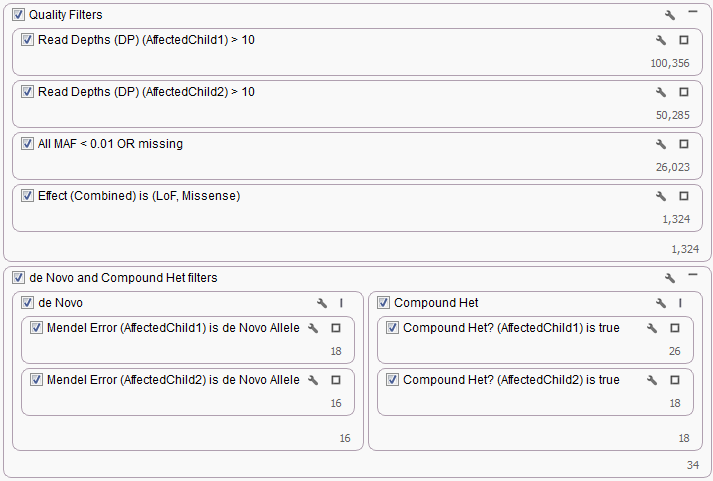
In most cases simply setting each filter card as sample specific and then duplicating the card to specify the second sample was enough to complete the workflow. This was true for the Quad workflow in terms of the Read Depth and Genotype Quality filters cards as well as the specific Mendel Error and Compound Het algorithm filter cards.
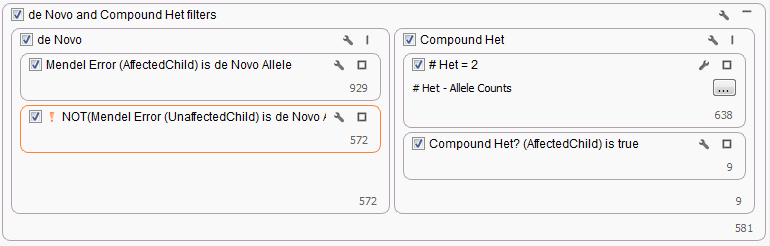
For other workflows, in addition to duplication of existing filter cards additional algorithms were required to finish the analysis. In particular, for the Trio plus Unaffected Sibling Workflow the Count Alleles algorithm was required to restrict the number of heterozygous variants to only 2, this would guarantee for any variant position only the single parent and the Proband would have a heterozygous call that could be used to determine the Compound Heterozygous status of the gene region.
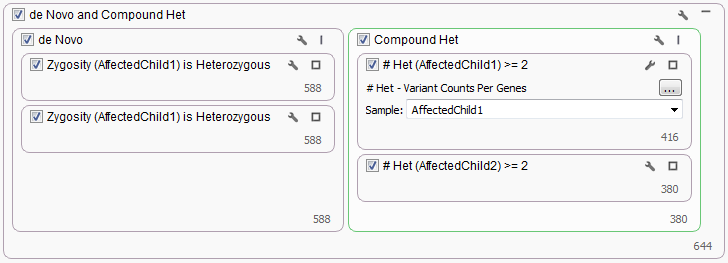
The last workflow looked at what would happen if there were only had two affected children available for a family. Since no parental genotypes were available we were not able to use either the Mendel Error or Compound Heterozygous algorithms to filter variants. Instead for the de Novo workflow, a Genotype Zygosity filter was used to find heterozygous variants in both affected children. Then the Count Allele by Gene algorithm was used to determine those gene regions where each child had at minimum two heterozygous genotypes.
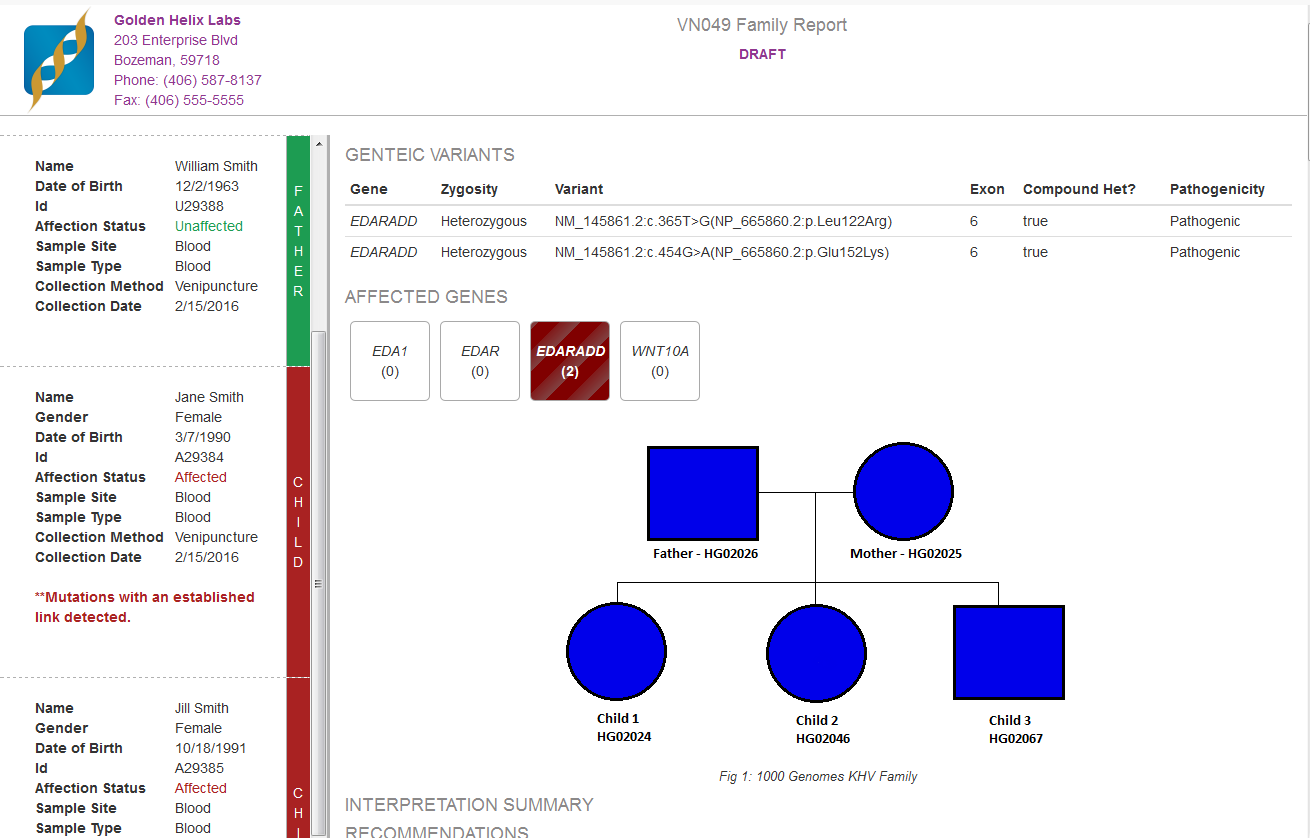
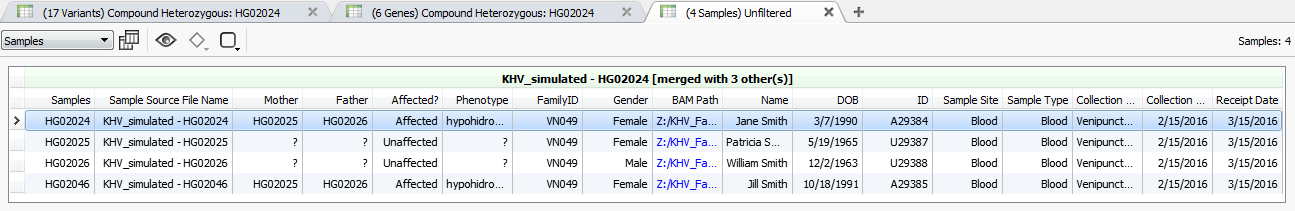
In some of the tools specific sample information was being auto-filled, for example, all of the patient information (Name, DOB, Sample Type, etc.) in the Quad Clinical Report, see Fig. 1, and the phenotype of interest for the PhoRank algorithm. This information was being grabbed automatically from the Samples table. Information in the Samples table must be specified during import using the From Text File option on the sample information dialog, see our manual at the following link for specifics on importing this data into your VarSeq project.
All of the templates that were created during the webcast are available for download from the following link. Once downloaded uncompress the folder and then from VarSeq go to Tools > Open Folder > User Data Folder and save a copy of each template in your Project Templates folder, they will then be available in your template list when creating a new project.
If you would like access to the customized Quad Report Template please email us at [email protected] and we would be happy to help you try it out.



