Our recent release of VarSeq 2.2.2 comes with a long list of upgrades and new features. In this blog post, we will demonstrate how defining sample phenotypes are available in VSClinical. One noticeable change is the ACMG guideline variant evaluation in VSClinical. Not only has this interface added CNV guideline evaluation, simplified the reporting process with embedded Microsoft Word and PDF rendering, but also now includes phenotypic capture for variant evaluation (Figure 1).
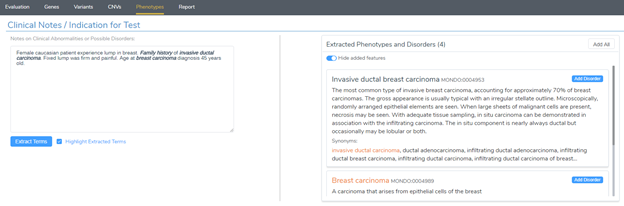
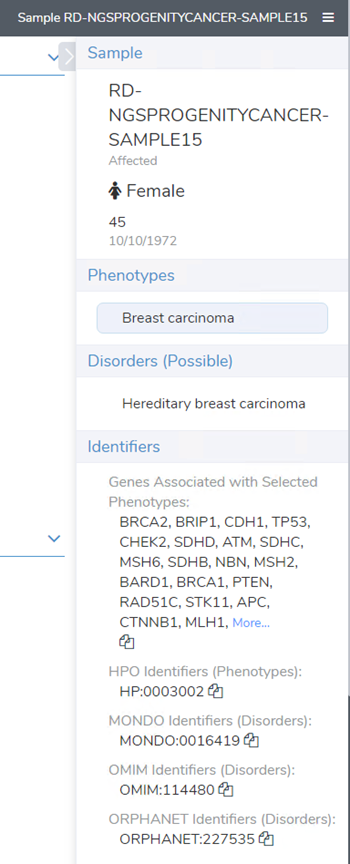
The utilization of patient phenotypes and disorders is incredibly effective in filtering variants with algorithms such as PhoRank. It is also necessary to include in the variant evaluation following ACMG and AMP guidelines in VSClinical, and ultimately the report. If the phenotypes are established in advance, they can be easily included in a sample manifest upon import and carried into the evaluation and report in an automated fashion (Figure 2).
However, if the phenotypes or disorders were not provided, users now have improved searching capability directly from the Phenotypes tab. You may have noticed the example in Figure 1 wherein a user can manually enter clinical notes, and then be parsed to extract key terms to compile the list of related phenotypes and disorders. Additionally, users can enter search terms in the Selected Phenotypes and Disorder section (Figure 3).
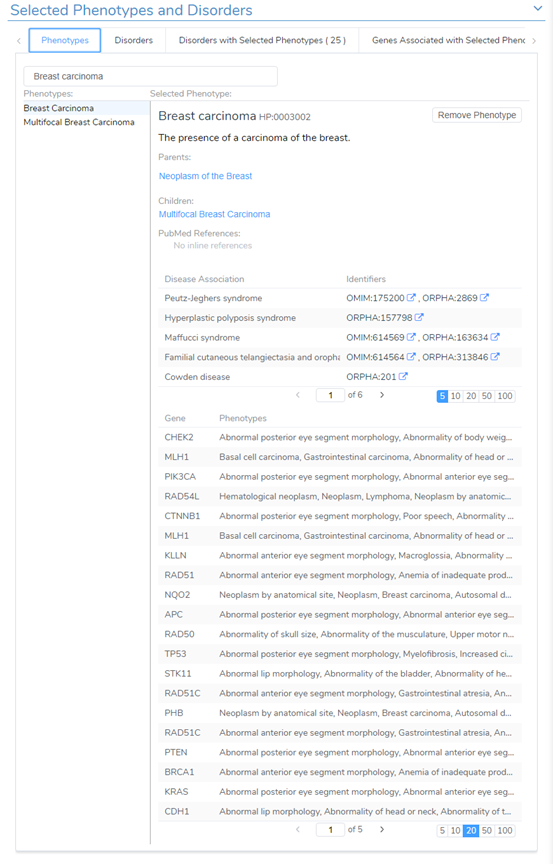
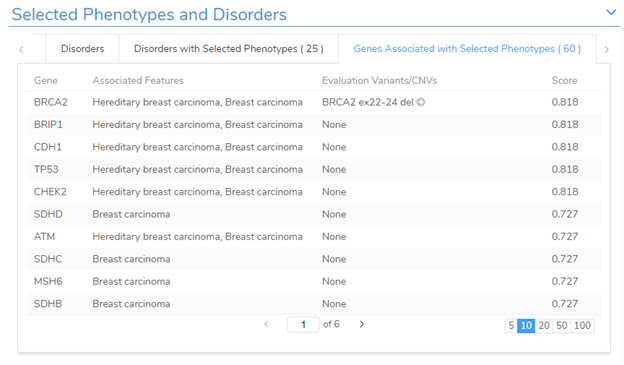
The phenotypic search will also include a listing of related genes with an association score. The score is multidimensional which incorporates disease-phenotypes curated across multiple databases, along with gene data from ClinGen, OMIM, ClinVar, GnomAD, and DECIPHER. The score indicates the gene dosage sensitivity in addition to how well the phenotypes match diseases associated with the gene. Additionally, this window will present you with previously recorded variants/CNVs evaluated in the past.
I hope you enjoyed reading this blog post on defining sample phenotypes inside VSClinical. If you would like 1:1 training on how to utilize this function or any other tools in VarSeq, please reach out to us at [email protected]. Feel free to also check out some of our other blogs that always contain important news and updates for the next-gen sequencing community.



