Genome-wide association studies (GWAS) are useful in genetics as they test for the association of a phenotype with common genetic variants. In contrast to the candidate gene approach, which is similar in practice, GWAS is “hypothesis-free” and does not require prior knowledge of a gene’s biological impact on a trait. The catch though is that this leads to analyzing hundreds to thousands of genome-wide array samples to elucidate single nucleotide polymorphisms (SNPs) associated with a specific phenotype. With enormous sample sizes required to validate findings, it is also necessary to have proper data quality assurance steps. To facilitate this process, the development team at Golden Helix strives to implement a few quality control and quality assurance steps in our SNP & Variation Suite (SVS) software that performs GWAS analysis.
Included in the first steps of quality assurance is filtering samples with low call rates and identifying samples whose inferred gender does not agree with their reported gender. The sample call rate is defined as the number of called SNPs per sample over the total number of SNPs in the entire dataset. A standard threshold for low call rate may be 95%, or in other words, if the number of SNPs called on an SNP array is below 95%, then the sample should be filtered out or re-evaluated. Using SVS, we can also assess inconsistencies between inferred and reported gender by visualizing heterozygosity and gender in a histogram plot. These initial steps allow the identification of low-quality samples, which can then be verified in the data and applied to your genotype spreadsheet.

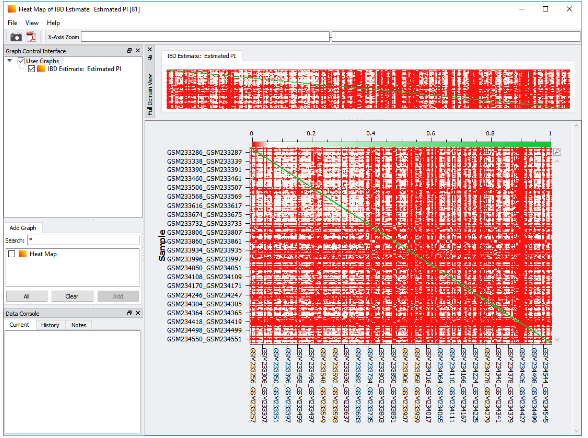
An additional quality assurance step would be filtering on cryptic relatedness. Cryptic relatedness is generally defined as a kinship among the cases or controls that could increase the false positive rates of the study. To filter out cryptic relatedness, we first prune SNPs that are in linkage disequilibrium with one another, i.e., SNPs that are in non-random association at different loci in the population. After LD pruning, we then can run identity by descent (IBD). IBD is a matching section of DNA shared by samples that have been inherited from a common ancestor and can be viewed in SVS using a heat map. The heat map can be useful for visualizing patterns of relatedness and can be evaluated for assessing first- and second-degree relatives, as well as identical twins. With this information, as well as the intent of your study, you can then remove sample pairs that are cryptically related or even 100% match duplicate samples.
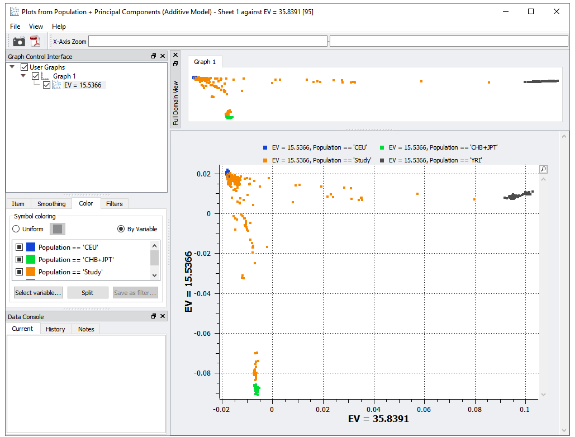
The third quality metric is to identify samples that depart from the expected homogeneity by evaluating the population stratification. Population stratification refers to differences in allele frequencies between subpopulations in a population and is typically the result of non-random mating between groups. Non-random mating is a problem for GWAS because the association to a phenotype could be the result of the structure of the population and not the disease-associated variant. In SVS, population stratification is assessed by performing principal component analysis (PCA). PCA can be performed by appending the filtered SNPs with HapMap data, which will produce principle component eigenvalues. The eigenvalues can then be used to explain the majority of the stratification and can be visualized using an XY scatter plot. The XY scatter plot allows the identification of outliers, which can then be removed to reduce the population stratification in the data.
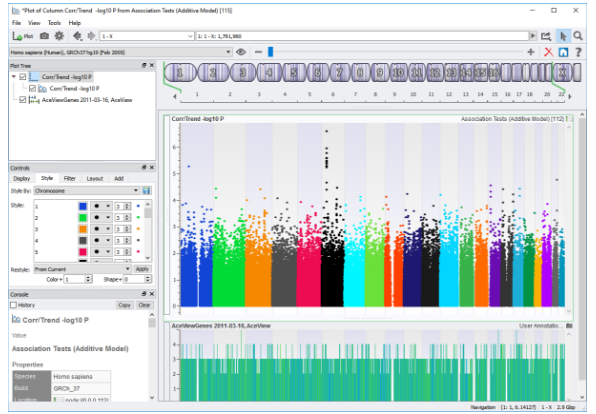
The last quality assurance metric applies to the SNPs. The two prominent metrics to filter SNPs in SVS are call rate and minor allele frequency (MAF). The thresholds for these metrics can be adjusted to your specific parameters but are generally set to drop SNPs if the call rate is below 90% and if the MAF is less than 1%. The MAF is the frequency of the less abundant allele at a genetic variant in a population and a MAF value of <1% is selecting for rare genetic variants. After these steps, association testing can be performed. Association tests in SVS include Genotype Association Analysis, creating Q-Q and P-Value plots, as well as creating Manhattan Plots.
Ultimately, these quality control and assurance steps are implemented to resolve many issues and limitations of GWAS. Common GWAS problems include lack of well-defined case and control groups, insufficient sample size, population stratification, and statistical problems resulting in false-positives. However, with these quality control and assurance metrics applied, GWAS can then be informative to some undiscovered variants that contribute to influencing a phenotype. As a result, there have been many scientific publications that have utilized SVS for GWAS. In 2018 alone, scientists have used SVS to identify genetic variants associated with non-small cell lung cancers, pathological misalignments of the eyes, Alzheimer’s disease, pain management for advanced cancers, and in multiple sclerosis to name a few. Additionally, SVS is used outside the realm of human genetics in fields including
In summary, there are multiple quality control and assurance steps that can be performed in SVS to help reduce the false-positive rates and optimize your GWAS analysis. To become more familiar with these techniques, feel free to look over our SVS tutorials and manuals and watch some of our webcasts on the specific topic. Otherwise, please feel free to comment in the section below or email support directly. We look forward to hearing from you.



