Thanks to all those who attended the recent webcast by Dr. Rana Smalling, “Integrating Custom Gene Panels for Variant Annotations”. If you were unable to attend or would like to recap, here is a link to watch the broadcast. We covered a lot of content regarding virtual gene panels, and there were several questions submitted during our Q&A session that were not fully addressed in the live session. So, the purpose of this blog is to provide answers to these questions about virtual gene panels.
When using VarSeq for any variant filtering workflow, ranging from small gene panels with a limited number of genes to whole-genome sequencing where you typically start with millions of genes, users typically employ standard filtering strategies for identifying clinically relevant variants. For the webcast, we highlighted how powerful our virtual gene panel tools can be for optimizing a variant filtering strategy (Figure 1). We approached the topic of using custom virtual gene panels from our users’ perspective, by covering several implementations that are becoming increasingly common among customers that use Golden Helix for their NGS analyses.
For the webcast, we used two clinical examples to demonstrate the usefulness of our gene panel tools. First, we used a batch of samples from a comprehensive cancer gene panel to demonstrate a per sample gene panel workflow, creating and managing panels using the Gene Panel Manager and investigating secondary findings from the ACMG Secondary Findings gene list. Our second example was a germline trio for an epileptic seizure disorder where whole-genome sequencing was performed on the two parents and the proband. We used this example to demonstrate how to create a template that is applicable for a whole-genome trio analysis and show how powerful the gene panel filter is for identifying relevant pathogenic variants in this context. Now, we will answer several questions from the Q&A session to provide more insights into the capabilities of VarSeq:
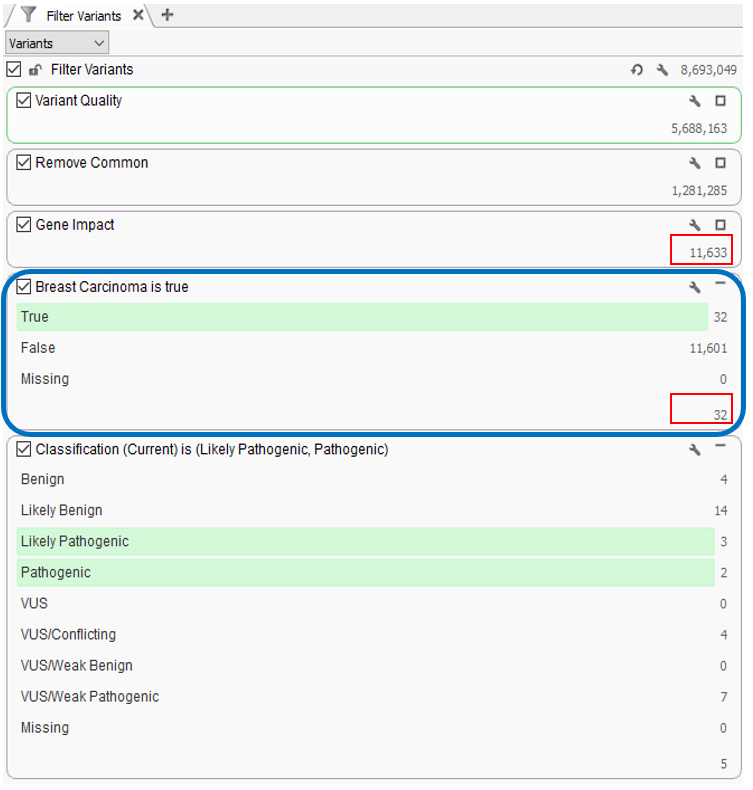
Can we leverage phenotypes or disorders instead of searching based on a gene panel?
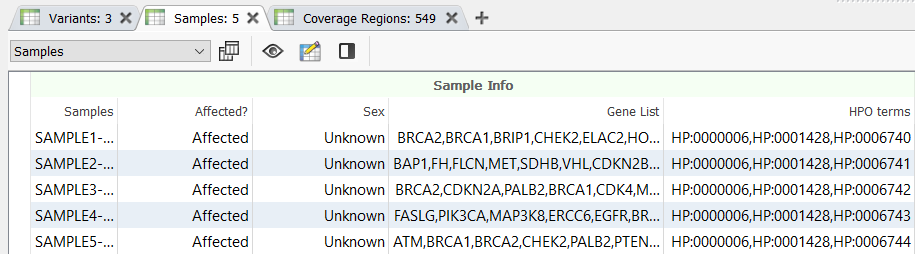
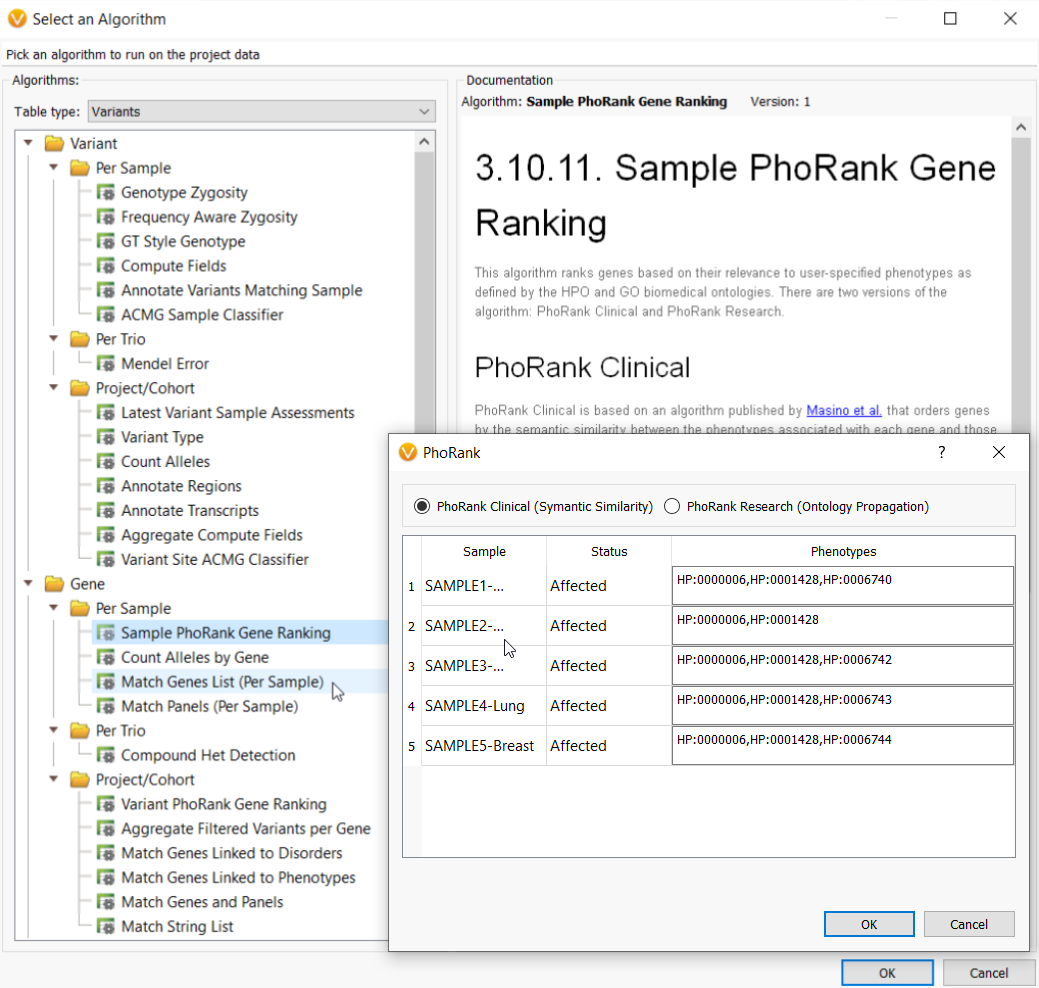
In the webcast, we showed users how to bring in a custom gene panel as a sample field using a text file manifest (Figure 2), and then utilize our Match Genes List algorithm (Figure 3) to annotate and filter based on a unique gene list for each sample in a batch or cohort. Similarly, you can leverage the text manifest to bring in specific HPO terms into your Samples table (Figure 2) and then utilize our PhoRank algorithm (Figure 3) to annotate and filter based on phenotypes. The HPO terms brought in will be automatically used to populate the list of phenotypes used by PhoRank to rank genes based on the relevance of the genes to those phenotypes defined by the user.
Can you use a bed file to define the panel?
We also showed users how to use their unique per sample gene panel to specify the list of genes for reporting NGS coverage in VSClinical. Similarly, you can use a bed file to define a gene panel. In the figure below (Figure 4), we see how in VSClinical there are several options for Reported Genes based on a gene panel. You can choose from the Sample Table “Gene List/Panels” or the list of genes in your Coverage Regions as defined by a bed file. The bed file is also used to define target regions for coverage calculation. Therefore, when using a bed file to define your panels it is important to keep in mind that as your panels evolve, you will need to update your bed file and recalculate target coverage regions to encompass the appropriate genes.

Can we subset the imported data to the panels in order to limit the number of variants in the project?
A sample specific gene list can be very useful when trying to narrow down the number of variants by filtering in a large scale analysis such as whole genome sequencing. Some users may even want to limit the number of variants to a gene list even earlier on import. An efficient way to accomplish this to use a bed file to define your target regions at import and subset the total variants to those targeted regions. You would do this by using the Subset to Track option, checking Import Regions Defined by Annotation File and selecting the desired bed file by clicking on Select Track (Figure 5).

We always appreciate these great questions from our community. For a deeper dive into any of these topics, please check out this webcast by our Director of Research, Dr. Nathan Fortier, or any of our other blogs and webcasts specific to the topic of virtual gene panels. As always, if you have any questions regarding the contents of this blog, please leave us a comment down below or reach out to us at [email protected].



