Thank you to those who attended our recent webcast, “PhoRank 2.0: Improved Phenotype-Based Gene Ranking in VarSeq”. For those who could not attend, you can find a link to the recording here. This webcast covered upcoming improvements to the PhoRank phenotype-based gene ranking algorithm based on literature published in the years since the algorithm’s development.
The PhoRank Algorithm
When performing variant analysis on whole exome sequencing data, clinicians must sort through thousands of variants to determine which variants are most likely to be associated with the patient’s phenotypes. To assist with this process, we have implemented the PhoRank algorithm, which incorporates phenotypic associations to highlight the most relevant genes with potentially damaging variants.
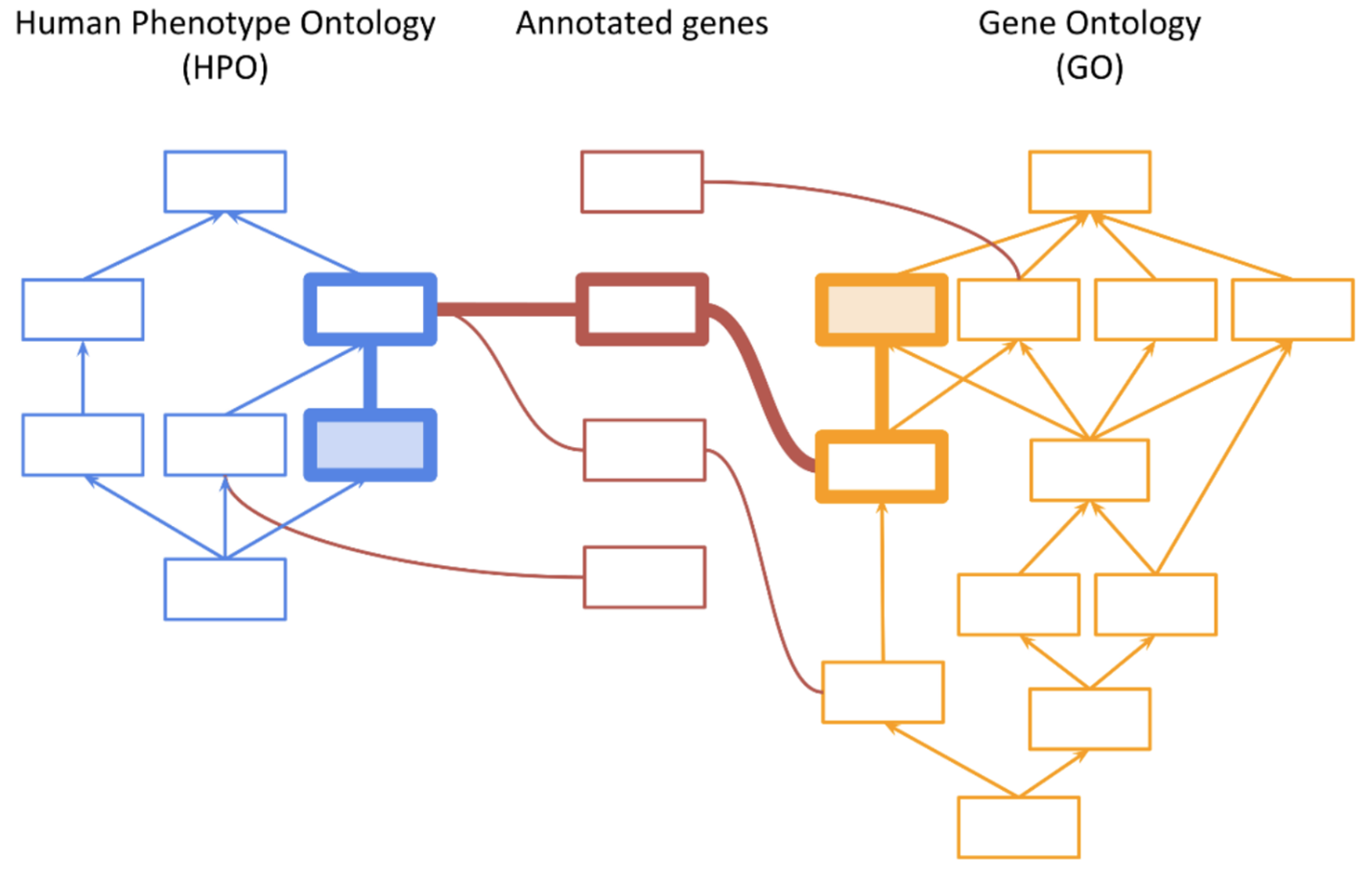
The initial implementation of the PhoRank algorithm in VarSeq was based on the methods utilized by the Phevor algorithm published by Singleton et al. [1] This approach utilizes a method called ontology propagation to combine information across multiple biomedical ontologies by leveraging shared gene associations between the ontologies, as shown in the figure below:
Innovations in Phenotype-Based Gene Ranking
Since the release of PhoRank, researchers have continued to innovate in the field of phenotype-based gene ranking. These advancements have inspired us to revisit the PhoRank algorithm and review the published literature to identify potential algorithm improvements. During this literature review, the following three algorithms stuck out to us, as they utilize methods that could potentially be used to improve our existing PhoRank algorithm:
- Phenolyzer: uses multiple gene-disease databases and ontologies in conjunction with a machine learning model to prioritize genes [2].
- Exomiser: combines ontology-based gene prioritization with model organism data and protein-protein interaction data [3].
- Masino’s Algorithm: orders genes by the semantic similarity between the phenotypes associated with each gene and those associated with the patient [4].
Benchmarking
We have compared these algorithms to evaluate the possibility of utilizing these methods in an updated PhoRank algorithm. This comparison was performed using a dataset developed by Pengelly et al. specifically for the purpose of benchmarking phenotype-gene ranking algorithms [5]. This dataset consists of 21 individuals with previously established clinically confirmed molecular diagnoses determined through traditional testing. Phenotypes for each individual were described through a comprehensive set of HPO terms, which were selected based upon a review of the clinical notes for each patient. We compared the ranking of the causal gene for each individual along with algorithm runtime across the four algorithms discussed above. The results of this comparison are shown in the table below:
| Phenolyzer | Exomizer | Masino | PhoRank 1.0 | |
| Average (min, max) | 453 (1, 6588) | 3461 (6, 6228) | 89 (1, 762) | 478 (7, 1729) |
| Samples with Gene in Top 10 | 6 | 2 | 9 | 1 |
| Runtime (minutes) | 38 | 5 | 16 | 84 |
For this benchmark dataset, Masino’s algorithm demonstrated superior performance in both average rankings of the causal gene and runtime. The competing approaches’ incorporation of additional gene-disease databases, model organism data, and protein-protein interaction data did not seem to provide any advantage on this benchmark dataset. However, it should be noted that the selected benchmark includes many phenotypes with well-established gene relationships, as would be expected in a clinical setting. We suspect that these more complex methods may be better suited to ranking genes without established phenotypic associations, but further testing would be needed to demonstrate this. Based on these results, we have chosen to use Masino’s algorithm as the basis for a new VarSeq algorithm that we have dubbed PhoRank Clinical.
ProRank Research and PhoRank Clinical
While PhoRank Research Excels at finding gene associations in individuals with atypical disease presentations, this method is less effective when applied to individuals presenting with disease phenotypes that have confirmed gene associations. The use of ontology propagation across multiple ontologies produces less optimal gene rankings compared to Masino’s algorithm when applied to phenotypes with well-established gene associations.
To address this, we have incorporated the methods used by Masino’s algorithm into PhoRank Clinical, a new phenotype ranking algorithm in VarSeq which utilizes semantic similarity to rank genes based on their phenotype associations in the Human Phenotype Ontology. Our empirical analysis demonstrates that this new algorithm provides better gene rankings when applied to phenotypes with established gene associations while producing results in a fraction of the time required by our existing PhoRank algorithm. This feature will be available in the next VarSeq release, which is slated for this Fall.
References
- Singleton, Marc V., et al. “Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families.” The American Journal of Human Genetics 94.4 (2014): 599-610.
- Yang, Hui, Peter N. Robinson, and Kai Wang. “Phenolyzer: phenotype-based prioritization of candidate genes for human diseases.” Nature methods 12.9 (2015): 841-843.
- Jacobsen, Julius OB, Damian Smedley, and Peter Robinson. “Exomiser and Genomiser.” Computational Exome and Genome Analysis. Chapman and Hall/CRC, 2017. 387-406.
- Masino, Aaron J., et al. “Clinical phenotype-based gene prioritization: an initial study using semantic similarity and the human phenotype ontology.” BMC bioinformatics 15.1 (2014): 1-11.
- Pengelly, Reuben J., et al. “Evaluating phenotype-driven approaches for genetic diagnoses from exomes in a clinical setting.” Scientific reports 7.1 (2017): 1-7.


