
Thank you to all our viewers who attended our webcast last week on VarSeq 2.5.0: VSClinical AMP Workflow from the User Perspective. Overall, this webcast highlighted the versatility of VarSeq, demonstrating both a Tumor-Normal somatic workflow and a singleton somatic analysis. In addition, we got to see the utility of our new cancer classifier and the upgrades to our Golden Helix CancerKB database. Read along for a recap of the highlights and several viewer questions we did not have time to answer while live.
Tumor-Normal Analysis Using VAF-Based Normal Subtraction
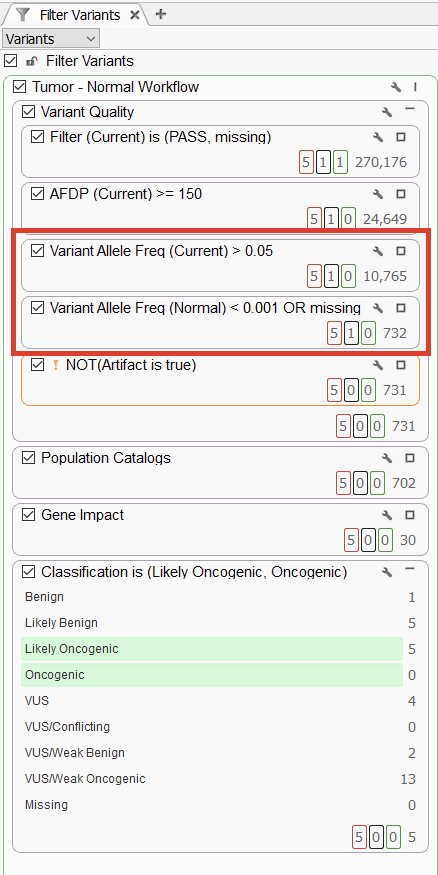
Our first showcased workflow was a tumor and normal project, where we used ‘normal subtraction’ to narrow the scope of variants to those unique to the tumor. This workflow demonstrates the ability of VarSeq to analyze variants from two VCFs at once to report on the shared outcome. This can be done in a filter chain by leveraging the variant allele frequency filters (VAF) (Figure 1). The first one is set to filter variants with a VAF of over 5% in the current sample, the tumor. The next card is set to filter for variants with a VAF of less than 0.01% or missing entirely from the normal sample.
In this workflow, we effectively filter from over 10,000 variants to just over 700 variants that are unique to the tumor. While this normal subtraction is very handy in the filter chain, we could leverage these filters in other ways, such as isolating secondary germline findings, which we will demonstrate with the next project.
Isolating Secondary Germline Variants with Parallel Filter Chains
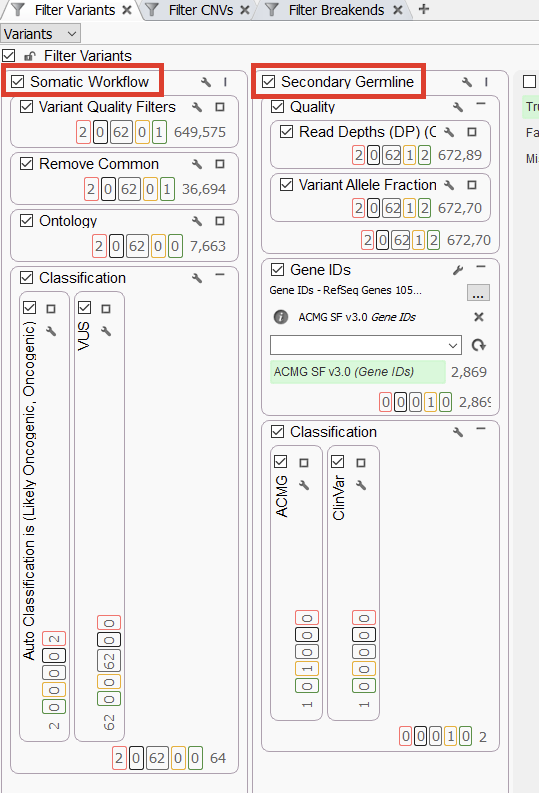
Our second project is set up to isolate secondary germline variants through the use of parallel filter chains (Figure 2). On the left, we have a somatic workflow, leveraging filters similar to the first workflow (Quality, population catalogs, and ontology). We leverage the Cancer Classifier for the last bin to look for Likely Oncogenic and Oncogenic variants. In parallel, we are filtering for variants that are VUS or VUS/ Weak Oncogenic, in addition to applying different Cancer Catalogs. The goal is to narrow the scope of remaining variants to those in genes that have known cancer associations. As described in the webcast, these variants would then be loaded into VSWarehouse for further assessment down the line.
The other major workflow here is the Secondary Germline filter chain. Like before, we filter for variant quality before applying a Gene Panel. The gene panel could be for anything, but here it is: the ACMG Secondary Findings gene panel. That reduces us to just less than 3,000 variants, down from the 672,000 variants in the above card. From there, we have our ACMG classifier for Pathogenic and Likely Pathogenic variants in parallel with a ClinVar pathogenic submission search. This gives us two variants that we can bring into VSClinical for further analysis and reporting.
Streamlining Somatic Workflows with the New Cancer Classifier
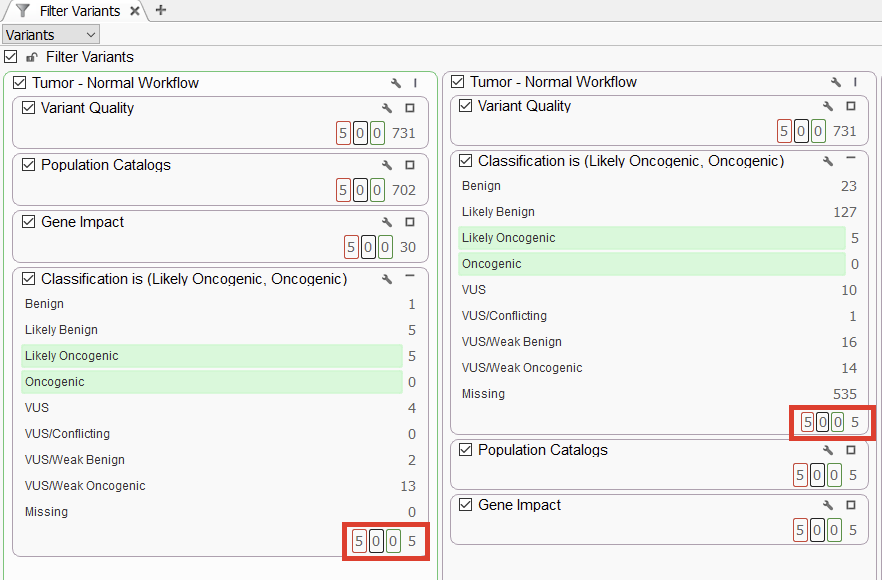
Our next major topic was our brand-new Cancer Classifier (check out the Cancer Classifier webcast for an in-depth discussion of how it works). Here, we wanted to showcase how well the Cancer Classifier can streamline a workflow (Figure 3). On the left, the Cancer Classifier is at the end of the filter chain, displaying five Oncogenic and Likely Oncogenic variants. On the right, we have moved the Cancer Classifier up above the population catalogs and the gene impact filters. What is amazing is that we still see those five variants isolated by the Cancer Classifier. This edition of the Cancer Classifier is a powerful addition to any somatic workflow.
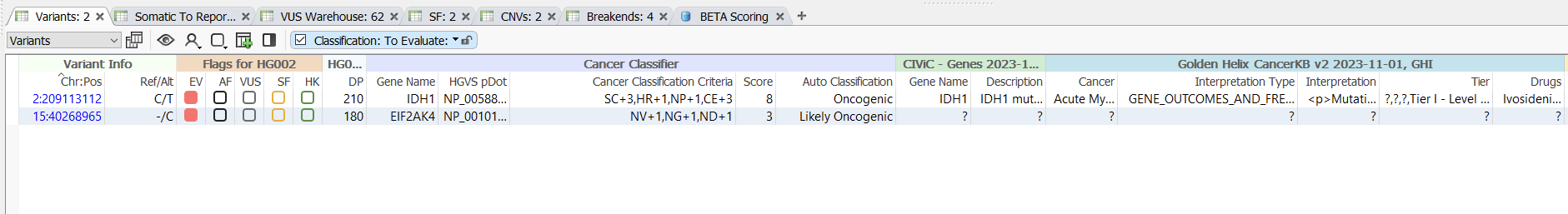
The Cancer Classifier can classify the oncogenicity of both variants that have been previously classified by a somatic database and novel variants, as seen below (Figure 4). The variant in IDH1 has been recorded across several databases, including CIViC and Golden HelixCancerKB, and comes in with an oncogenic classification. The EIF2AK4 variant has not been classified by either of these databases, but due to the accumulation of other evidence sources is still classified as Likely Oncogenic.
Introducing the New Golden Helix CancerKB Gene Track for Enhanced Cancer Insights
Our latest addition to the Golden Helix CancerKB family is the new Golden Helix CancerKB gene track! This gene track can bring in different information fields, such as the roles in cancer and cancer hallmarks.
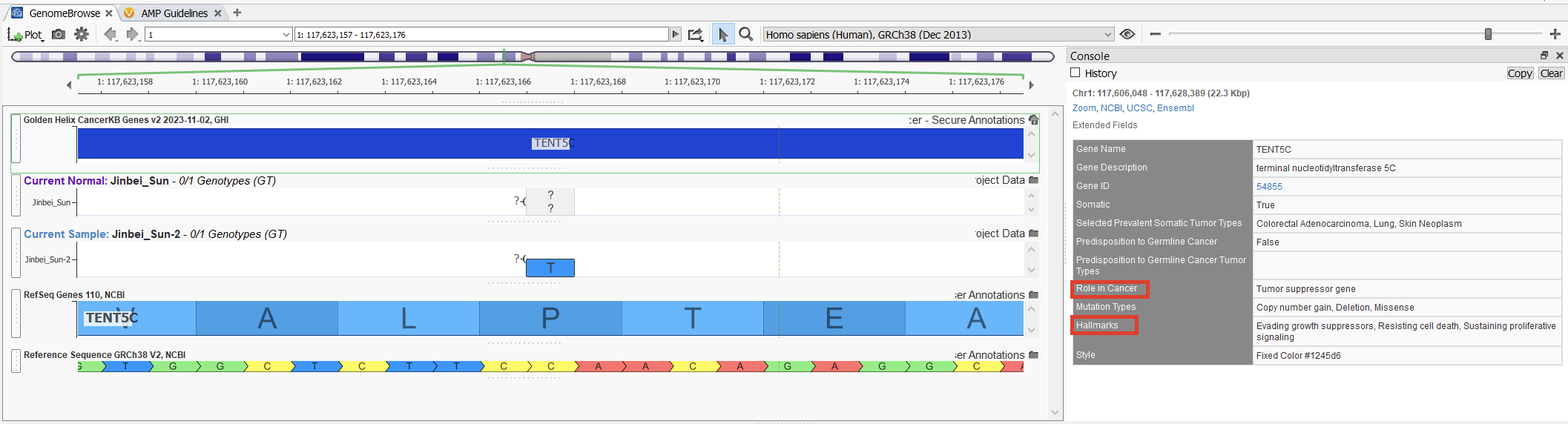
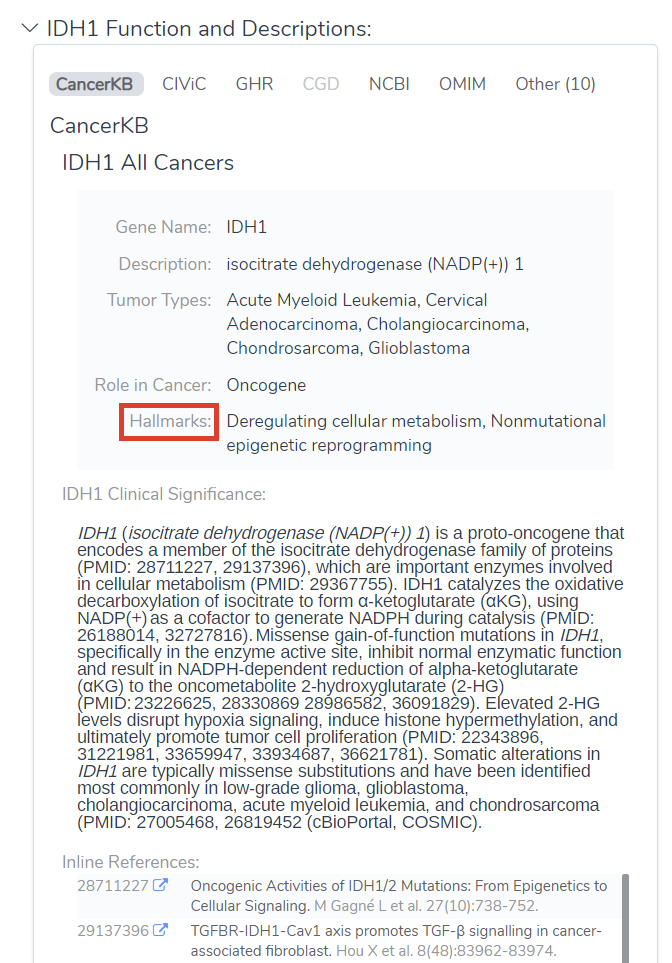
The cancer roles and hallmarks are now also included in the clinical significance section, as seen below (Figure 6).
User Questions, GHI Answers
Thank you again to our viewers who came to the webcast. There were some really good questions, and we wanted to answer a few we did not have time to go over live. To start, we had two Golden HelixCancerKB questions that are best answered together.
1. A. Can I make a change to a Golden Helix CancerKB Summary?
B. I’ve been adding more information to my biomarker interpretations that I think could help other groups- can I share those?
Absolutely! When viewing the biomarker summary, you can edit and interpret or write a new summary from scratch (Figure 7A).
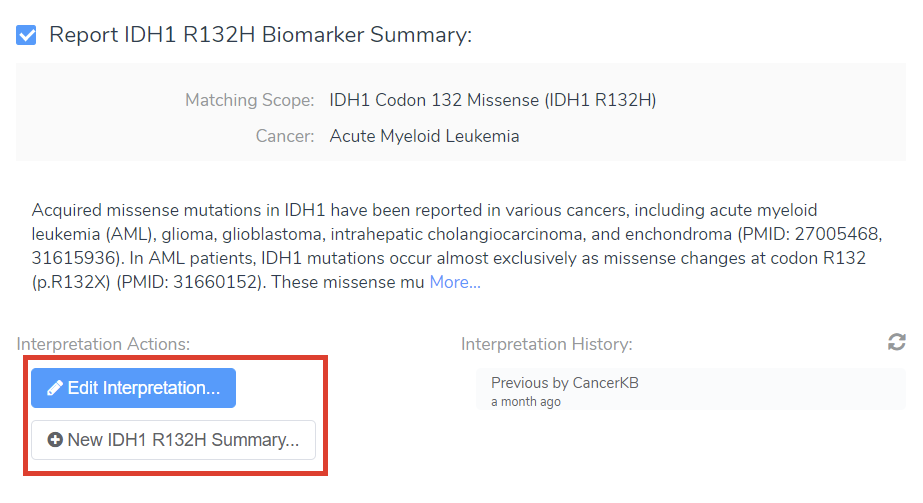
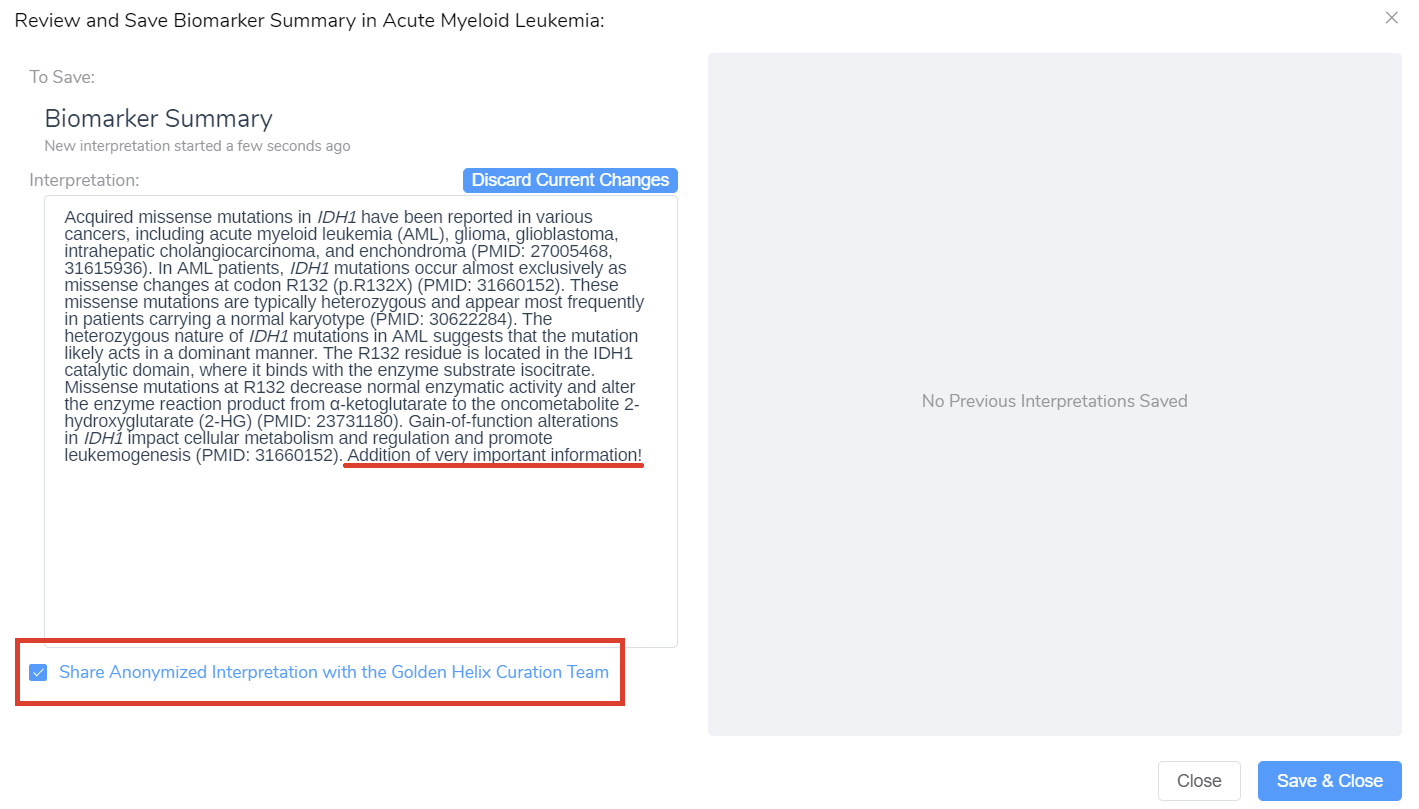
While inside the editing dialog, you also have the option of sharing those new interpretations with the Golden Helix CancerKB Curation Team (Figure 7B). The summary will be reviewed by a cancer professional and then possibly added to the growing Golden Helix CancerKB database.
2. Can the Cancer Classifier score variants that have not been seen before?
This point was so important we decided to feature it twice (see Figure 4). Here in Figure 8, we have an example of a variant that has been previously classified by a number of cancer catalogs and is being classified as Oncogenic. We also have a novel variant that has not been seen by these databases. Due to the accumulation of knowledge from other sources (null-variants in the oncogenic gene, nearby pathogenic variant, and loss of function in the oncogenic gene), we are able to classify this as a likely oncogenic variant.

3. Can we implement new evaluation scripts into VSPipeline workflows?
Yes! VSPipeline does support running evaluation scripts to give you the most streamlined automated project creation possible.
In summary:
Our webcast covered some of the highlights from the VarSeq 2.5.0 release. We explored two different somatic workflows, one for a tumor and normal pair, the other a single tumor sample. In addition, we explored the power of the new Cancer Classifier. We also looked at how Golden Helix CancerKB can be used to streamline oncogenic exploration.
Thank you for coming, and if you have any questions about how these new features can be used in your workflows, please contact our team at [email protected].



