
Phased genotypes provide crucial information that allows us to separate variants into distinct haplotypes, representing the sequence of alleles inherited together from a single parent. This information can offer profound insights into inheritance patterns, the combined functional effects of variants, and the identification of specific genetic profiles such as pharmacogenomic diplotypes — all of which are central to effective genomic interpretation. In this blog post we describe how phased genotypes are represented in Variant Call Format (VCF) files, and explore the many ways this data can be leveraged during variant analysis.
Representing Phased Genotypes in VCF Files
In the VCF file format, phased genotypes are represented using a vertical bar ‘|’ to separate the alleles (as opposed to the forward slash ‘/’ used to represent unphased variants). Phased genotypes should be accompanied by a phase set (PS) field, which defines a set of genotypes for which relative phasing is known. Phased genotypes that are on the same chromosome and share the same PS value are considered in-phase, meaning that the corresponding alleles were inherited together. If two variants share a phase set, but have genotypes that are on different chromosomes (e.g. 0|1 and 1|0) then these variants are considered out-of-phase. If two variants belong to different phase sets, then their relative phasing cannot be determined. The recommended convention for the PS field is to use the position of the first variant in the phase set as the PS identifier, though this is not required.
Joint Effects of In-Phase Variants
Phased genotypes allow for the analysis of the joint effects of multiple variants that are inherited together. In many cases, the impact of several variants on the same chromosome can differ significantly from their individual effects. Such differences can often be detected even in short-read data, where phased genotypes of nearby variants may reveal substantial joint effects. For example, consider the phased variants shown below:

Independently, these two missense variants might have uncertain effects on protein function. However, when their combined impact is assessed, it becomes clear that these mutations result in a stop-gain, likely resulting in nonsense-mediated decay.
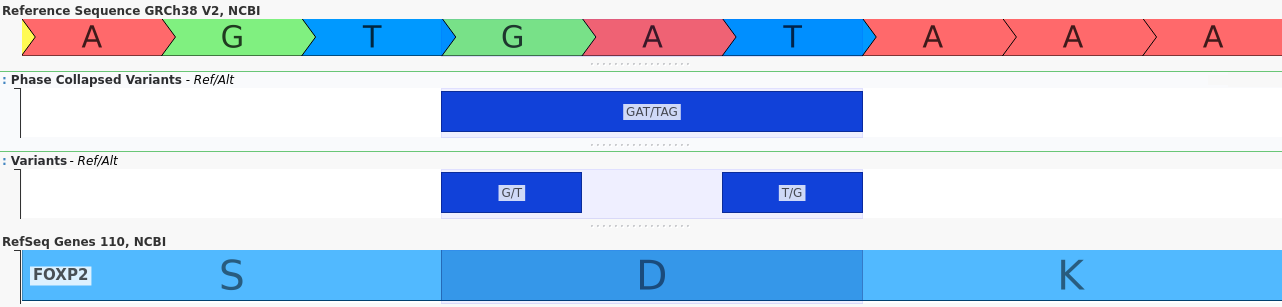
Long-read sequencing data further enhances our ability to analyze joint variant effects by providing phasing information for distant variants, including insertions and deletions. For example, the two variants shown below are separated by hundreds of base pairs:

When considered independently, both of these variants are frameshift mutations which are likely to have a deleterious effect. However, when their joint effect is considered, it becomes clear that the reading frame is preserved, mitigating their impact.
Inheritance Analysis
Phased genotypes are also useful in the analysis of inheritance patterns, especially when parental sequencing data is unavailable. One example of this is the detection of compound heterozygous variants. A compound heterozygous variant occurs when an individual inherits two different variants in the same gene—one from each parent—potentially compromising both copies of the gene. When two heterozygous variants within the same gene belong to the same phase set and have out-of-phase genotype values, it strongly suggests the presence of a compound heterozygous variant.
Differentiating Diplotypes in Pharmacogenomic Genes
Pharmacogenomics often relies on identifying specific diplotypes—combinations of haplotypes that determine the overall genetic profile of a gene. Phased genotypes are crucial in this context because they enable the accurate differentiation between potentially ambiguous diplotypes that may have identical unphased genotypes. This is important, as two different ambiguous diplotypes may result very different metabolizer phenotypes for certain drugs. For example, consider the three CYP2D6 variants in the plot below:
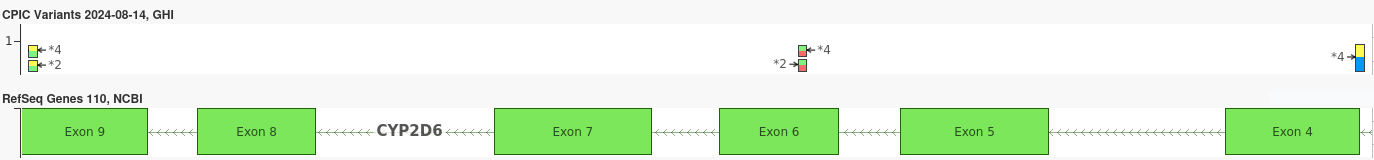
The two variants on the left are consistent with both the *2 and *4 alleles, while the variant on the right is the core variant which defines the *4 allele. If all three of the above variants were in-phase then we would assign the diplotype CYP2D6 *1/*4. However, if the *2 variants were known to be out-of-phase with the *4 variants, we would instead assign the diplotype *2/*4. In the case where phasing information is unavailable, it would be impossible to differentiate between these two diplotypes.
Conclusion
Phased genotypes provide essential information that enhances our understanding of genetic variants and their implications. From analyzing the joint effects of in-phase variants to differentiating between complex pharmacogenomic diplotypes, phasing data is invaluable in both research and clinical settings. In future blog posts we will dive deeper into this topic and discuss new VarSeq features for incorporating phased genotypes into your variant interpretation workflows. If you are interested in adding VarSeq to your NGS analysis, click the button below and start your evaluation.



