VarSeq now supports analysis of paired Tumor/Normal samples! Tumor/Normal support has been one of the most common feature requests for VarSeq since it was launched late last year, and we are excited to make this functionality available to all of our VarSeq users in the latest update (version 1.1.4).
VarSeq is a powerful platform for annotation and filtering of DNA sequence variants from many different study designs. It can handle data of any scale, from targeted sequencing to whole exomes and whole genomes. The new Tumor/Normal support creates a specific mode for handling paired samples. In this mode, the basic analysis unit is not one sample, but a pair of samples. Data filters can be created with decision rules specific to either the tumor or normal specimen, and those filters can then be saved as a workflow template that can be applied to large batches of samples in the future.
Previous versions of VarSeq supported analysis of sequencing data from three standard study designs selected by the user during data import: unrelated samples, families and cancer samples (usually tumor-only sequencing from gene panels). Beginning with VarSeq version 1.1.4, the list of available study designs includes Tumor/Normal, as seen in Figure 1.
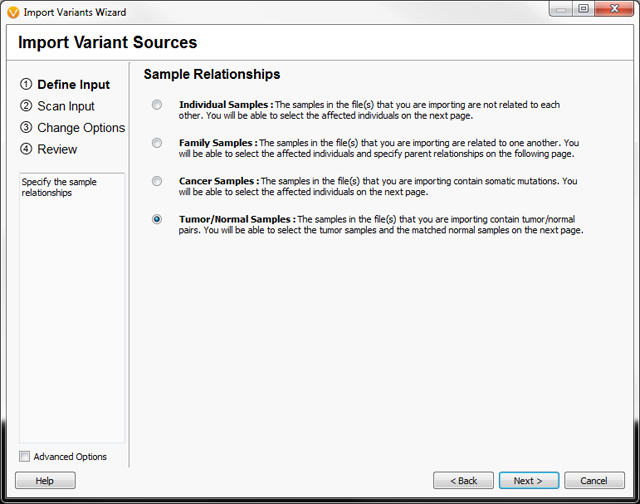
Figure 1: The VarSeq data import wizard now allows users to specify Tumor/Normal Study designs.
One or many sample pairs may be analyzed in a VarSeq project. The import wizard simply requires the user to specify which samples are from tumor tissue, and which sample is the matched normal for any given tumor, as shown in Figure 2. If there are many sample pairs, the user may also upload a text file with the matching information.

Figure 2: Using the VarSeq import wizard to specify sample pairs. A “Normal Sample” must be selected for any sample that is identified as “Tumor.”
VarSeq’s new Tumor/Normal mode incorporates features of the existing framework for cancer samples, including multi-allelic splitting during the data import process. This means that if more than one alternate allele is reported at any locus in the VCF file, VarSeq creates a record for each Ref/Alt pair. This allows the user to uniquely annotate each potential somatic mutation and ensures a fair comparison of alleles within the sample pair.
As with other study designs, analysis of Tumor/Normal pairs in VarSeq begins from VCF files. We suggest using a somatic variant caller such as MuTect or Somatic Sniper that will generate multi-sample VCF files containing technical data from each sample for every variant site. This ensures that you know exactly what is observed in the normal sample at every variant site reported for the tumor sample. VarSeq also allows you to visualize BAM files to review sequence alignments manually to confirm the evidence for any given variant (see Figure 3).
The Tumor/Normal mode is also fully supported in VSPipeline, making it easy to integrate in automated laboratory processes. A simple text file is required by VSPipeline to define the sample pairs within a batch of samples.
We hope that you will enjoy this new feature in VarSeq, and look forward to your feedback!
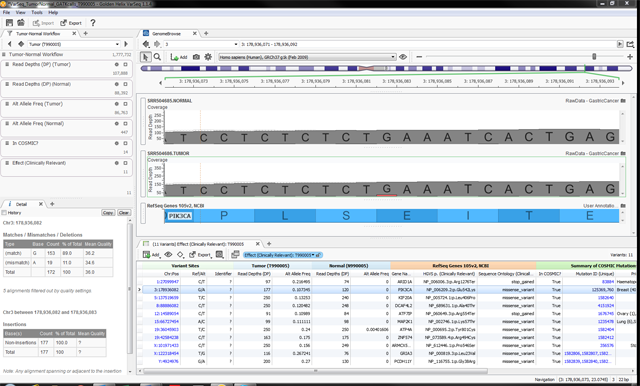
Figure 3: An example VarSeq project with whole-exome Tumor/Normal sequencing data. Variant data for the two DNA samples and related annotations are shown in the table at the bottom-right. A mutation in PIK3CA is highlighted. The GenomeBrowse window at the top shows sequence pileup data for the same mutation; the mutant allele is present in about 11% of sequence reads from the tumor. The filter controls in the upper-left are configured to identify potential somatic variants based on read depth and allelic fraction thresholds from the respective samples.



