Recently, I have been thinking a lot about Human Genome Variation Society (HGVS) notation — you know “G dot”, “P dot”, and “C dot”. HGVS has quickly become one of the most common ways to represent variants. It’s no wonder that HGVS nomenclature is used so widely. It provides an easily readable, compact representation of a variant. Since it is so prevalent, it’s helpful to take a step back and consider the scenarios where using HGVS nomenclature succeeds and where it fails.
The first version of HGVS was published in 2000 and really preceded the establishment of a human reference sequence. Thus, the nomenclature has the very desirable feature that it can describe variants (in C dot and P dot form) with no knowledge of the reference sequence–only the RNA or amino acid sequence must be known. For some portions of the genome we are lucky enough to have a stable coordinate system called a Locus Reference Genomic (LRG) segment. However, if the variant being examined is outside an LRG the notation is once again implicitly dependent on the reference genome. As I previously wrote, if the transcript is mis-mapped, the C dot notation will be incorrect as well.
The goal of HGVS notation is simply to communicate a variant using “consistent gene mutation nomenclature”. This goal is really twofold: communication and consistency. Judging by its widespread use, the communication that HGVS enables is a resounding success. Just like other forms of expression, we sometimes have to use the surrounding context to resolve ambiguities. But, usually the HGVS representation is specific enough to understand what the research or clinic report is talking about.
Bioinformaticians sometimes like to groan about the lack of specificity in the HGVS spec, but I actually think that they have done a fantastic job given the goal of the nomenclature. It has a well-defined core set of rules to construct an easily readable representation of a variant. That is not to say that the spec doesn’t have some problems which ultimately hamper the consistency of representation.
The spec doesn’t provide a unique representation for variants. We previously wrote about this failing, so I won’t spend too much time belaboring the point. However, you must be aware of this fact and not try to use HGVS to resolve variants into things like database keys or use it as a built in unique ID for variants. If you try to use it as a format suitable for machine processing you are begging for trouble, especially if representations are being generated by two separate libraries.
Before you notate: Look left, right, then left again
The community also seems to largely ignore or modify portions of the spec. These modifications don’t necessarily impede the end goal of HGVS, but they do beg the question of how the spec should evolve.
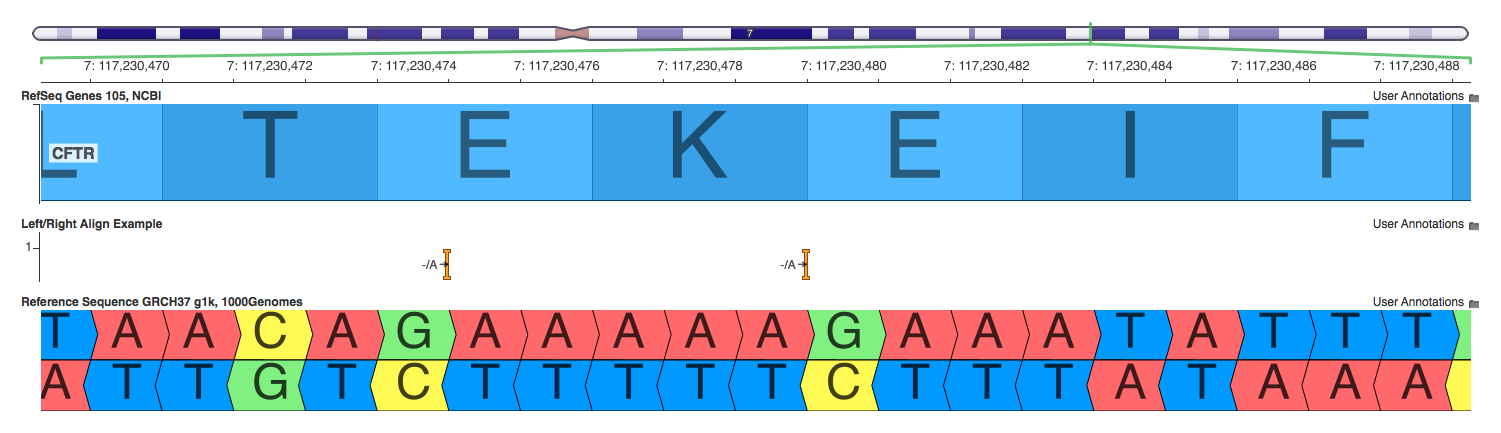
An important deviation often seen in HGVS’s representation is the alignment or justification of variants. The bioinformatics community has increasingly started to agree to left align variants when represented against the reference sequence. However, the HGVS spec urges “for all descriptions [to use] the most 3′ position possible,” which implies right shifting the variant on forward strand genes. So this means that our VCF has variants in arbitrary positions, our databases have left aligned variants, and our HGVS representation is right aligned (or maybe left aligned). This is madness! It is crazy to try to mentally left shift a variant when we read an HGVS representation and want to quickly cross reference with a different resource. Maybe due to this difficulty, I often see the HGVS conversions neglecting to shift or normalize the variant. In practice, it doesn’t matter all that much – the variant is still as correct as we can infer from the raw read data. Where it does pose problems is if you rely on the notation as a unique representation as discussed above.
As an exercise, I plugged in the two insertions from the screenshot above into a few common variant annotation tools to see how they would report each variant.
|
Left Variant |
|||
|
G dot |
C dot |
P dot |
|
| NCBI Variation Reporter | g.117230474_117230475insA | c.1747_1748insA | p.Glu585Argfs |
| VEP | N/A | c.1747_1748insA | p.Glu585ArgfsTer4 |
| SnpEff | N/A | c.1748*>+A | p.Glu583X |
|
Right Variant |
|||
|
G dot |
C dot |
P dot |
|
| NCBI Variation Reporter | g.117230479_117230480insA | c.1752_1753insA | p.Glu585Argfs |
| VEP | N/A | c.1752dupA | p.Glu585ArgfsTer4 |
| SnpEff | N/A | c.1753*>+A | p.Glu585X |
If the functional product of the gene is important, it’s probably better to use P dot notation. Unless you have the entire human genome memorized (I’m working on it…3 billion bases to go) seeing the C dot representation of a SNP adds little to the discussion. C dot won’t tell you if it is synonymous or nonsynonymous. It’s just position gibberish. P dot on the other hand at least gives you some idea of what’s going on at the functional level. It’s much more interesting to note that a Valine got turned into a Glycine (changing its sidechain from hydrophobic to hydrophilic) than that an exonic T was changed to a G.The main thing to note in these tables are the many representations of the same variant. For example, Variation Reporter is not right aligning in G and C dot cases, but is in the P dot cases. Similarly VEP doesn’t 3’ prime shift its C dot notation, but does for P dot. And SnpEff is not shifting either C or P dot. Aside, from the shifting issues, note how different each tool’s HGVS output is when attempting to annotate the same variant.
Considering our options
Even if we can get beyond these shortcomings, we still need to be cognizant of what style of HGVS notation we choose to use. It seems that by default most variants are represented using C dot notation. In many cases, especially where software is involved, C dot is a poor form on which to rely. The reason is that C dot introduces a lot of unnecessary ambiguity without a significant gain in informative context.
Your other choice would be to represent the variant using G dot notation. The aspects that support this choice are that it’s a relatively straightforward encoding and less prone to error than C or P dot. Moreover, assuming the same reference sequence there are many fewer ambiguous variants. By using G dot you’ve traded a few of the contextual clues (exonic/intronic/how many bases exonic) about a variant that C dot provides for the sound specificity of G dot. A worthy trade in my opinion, especially considering that a reader could type the G dot representation (almost without modification) into a genome browser and have all the context that C dot provides and more.
C dot run
We often split the difference and use C dot. Yes, we do gain some information, but this information can also be less cryptically encoded in plain English. Moreover, the idea that it will somehow span the communication gap among disparate researchers is empirically false. Depending on the tool used to generate your notation, the representation will vary enough to not be directly comparable (as seen above in our very simple insertion test case). Instead, we’re left in a situation where we need to translate two variants back to genomic coordinates (on the same reference sequence), shift both variants in the same direction and then compare them.
The case is often made that using C dot helps variants stand the test of time. As I touched upon at the beginning of this post, this is only true if we are in a region that has stable genomic coordinates (an LRG). In fact, HGVS recognizes this fact as prefers the use of an LRG as the accession.
So, while in theory C dot will provide very consistent notation for variants across research groups and time, in practice it doesn’t. We see the use of different accessions, multiple representations of a variant, and find that to rely on the representation we need to know quite a bit about the sequence underlying the transcript. In fact, one 2011 study found that almost 25% of C dot notations were incorrect. Thus, the whole idea of a unified, consistent representation has been lost to complexity.
C dot may be the notation of choice when we really care about pre-spliced RNA or in well-defined clinically significant regions with a LRGs. But, for widespread consumption I’d say it is better to use the simpler G dot or the more informative P dot notation plus some English and a screenshot. If the list of variants being described is large enough to preclude this, it might be time to attach a VCF.
Perhaps the best critique is that with our increasing dependence on software to manage the tsunami of variants that come with every sequencing run — a challenge central to NGS data analysis, we need to work on ways to uniquely identify variants in a machine-readable way. Accurate variant notation is a foundational step in genomic interpretation, where ambiguous representations can cascade into downstream errors. A 25% error rate is simply unacceptable when dealing with data. Currently, LRGs, VCFs, and lift-overs are part of the solution. I’d argue that an open sourced reference implementation of HGVS is another part. However, until we see that implementation, variant database curators will still be saying “send us your VCF” and not “send us your C dot.”
Ultimately, I’m a realist and I realize that it is unlikely for the momentum behind the use of C dot notation to wane, but I hope that you’ll stop and think about what you are trying to communicate and the associated trade-offs with each notation style. By being aware of some of the underlying assumptions of something as seemingly simple as variant notation we can avoid common nomenclature pitfalls.




Hi Andrew, great post. I have been working on a fork of SnpEff (https://github.com/CBMi-BiG/snpEff) which attempts to arrive at “canonical” coding HGVS by performing a “walk-and-roll” of indels in the 3′ direction. We expect the official SnpEff will adopt this strategy in the near future.
Glad you enjoyed it!
I’ve actually looked at your repo and it looks good! It’s no easy task to 3′ shift everything!
It’s amazing to see the amount of inconsistency between HGVS notation both from edge cases in the spec and from different implementations. It’s why I think having a reference implementation would be a great thing, but until that happens users will just have to proceed with caution.
Really interesting post.
I have to agree that g dot and p dot are the most informative and useful forms of the notation.
c dot notation is a pain even in clinically relevant regions. It is completely dependent on the context of the transcript of interest. For a typical human gene with multiple transcripts the c dot notation can be very different, plus there’s no requirement to specify *which* transcript you are refering to. It’s common to see GeneX c.1224delA where the gene has >1 transcripts.
p dot notation also suffers from this, as each transcript can code for different protein forms, but at least you can still see that you’ve introduced a permature STOP or something. Not so with c dot.
Great Point! It’s definitely important to consider the way this notation is used in the real world.
Excellent post.
Just to let you know that the VEP can now 3′ shift HGVS c notations using the –shift_hgvs flag.
We’ve decided to make it optional rather than the default as we think it will confuse users if we change the position of their variant from the input! Then again, we probably confuse other users by not producing the exact HGVS spec…
Good to know! It’s confusing no matter how you slice it. But, generally I agree that modifying people’s input can be confusing. If nothing else, it adds a second or two of “Wait, what happened…Oh I see.” And these little delays can certainly add up (both in time and cognitive load)!
Thanks a lot, very interesting post. However, I do not understand why you did not refer to the annotator built by the HGVS, MUTALYZER, which is the final word on the subject. Any genetics journal will ask you to name your variants according to Mutalyzer, available at http://www.mutalyzer.nl.
this is a learning experience for me . My knowledge is limited to SNPs and Sanger Sequencing. So can you Guys educate me on this topic on HGVS . I am aware of QRT-PCR and also microRNAs. but havn’t had an opportunity for whole genome sequencing.
this article was very interesting to me hence eager to learn more – and eager to see how I can use it for my patients’ samples.
best regards
Kaiser Jamil