The Beginning of Your Tertiary Analysis
VarSeq is designed to be your NGS tertiary analysis solution providing users simple but in-depth means of exploring gene panel, exome, and whole genome variants. For those not accustomed to the VarSeq software, the main import file for variant analysis is the VCF. Those who are familiar with the VCF know that there can be many flavors of specific fields for the variants and samples, but for the most part, the overall file structure is consistent. You can see the common VCF format descriptions here. Typically, the VCF format is determined through the secondary analysis process, and each secondary tool may be unique across users. One major concern might be, how do I know VarSeq can handle importing my VCF? The purpose of this blog is exposing the readers to some key elements of VarSeq’s universal VCF importer.
One first concern may be that you may not know what format your VCF has. VCFs can always be opened in notepad or some other text editor, but sometimes the number of variants is too large for the text editor to handle. Using VarSeq’s Data Source Library, users can Browse to their VCF file and scrolling to the bottom of the Information panel users will see the Info and Format fields defined in their VCF (Figure 1).
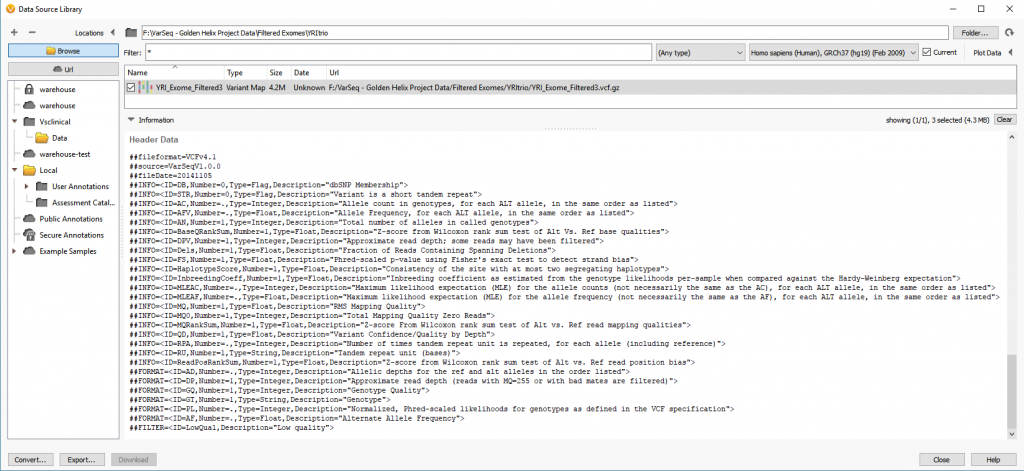
The info fields (Figure 2) in the VCF are specific to the variant itself while the Format fields (Figure 3) are data specific to a variant in a sample. The main thing to address here is that VarSeq will import everything that is present in the VCF file. In some cases, a VCF may have the sample-specific data represented in the Info fields. This is typically the result of performing the variant calling on a single sample VCF which doesn’t require the Format fields since every variant in the VCF is specific for the individual sample. In any case, the VarSeq importer allows for field format customization with advanced import.
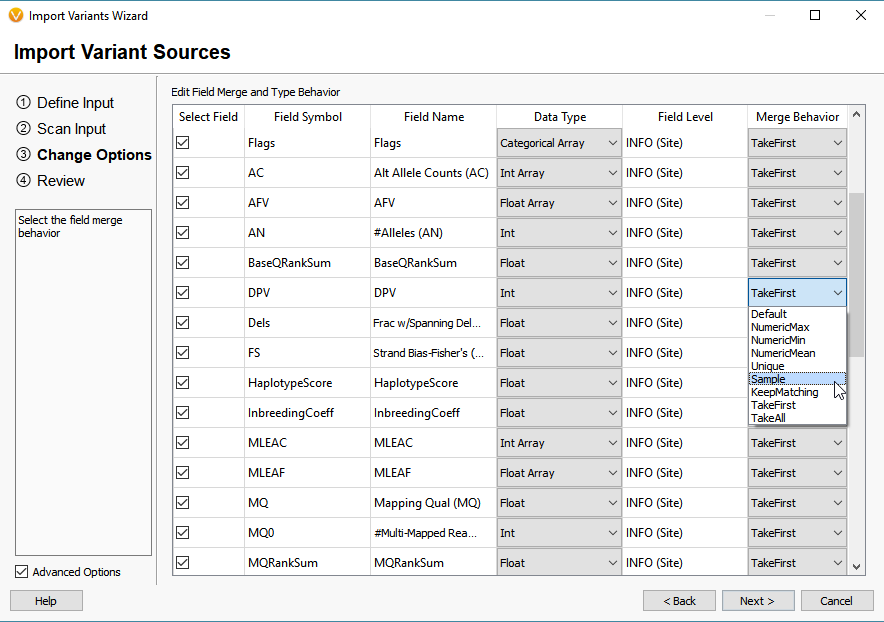
Through the Advanced Options, users can manipulate the original fields in the VCF and define the ideal merge behavior (Figure 4). This is important regarding the project templates users build in VarSeq. The project templates serve to define a workflow for filtering variants based not only on the quality fields in the VCF but also the long list of annotations and algorithms we provide. For example, if a workflow is built to filter variants based on the Read Depth (DP) Format field (Sample specific), modifying the merge behavior to change an Info Field to Sample may be necessary so to fit the standards of the filter workflow. If you are experiencing any troubles with filter workflow and VCF formats, our expert support staff can assist with defining the structure of the VCF and project workflow design. Please contact us at [email protected].
Another advanced import feature is to modify the variant normalization process (Figure 5). Fundamentally, VarSeq will utilize these normalization options so to represent the variant in its purest form and optimize it for variant annotation. There are a couple of steps here that are important for variant normalization. The first being left aligning the variants of which the Smith-Waterman algorithm will left align insertions and deletions for a canonical representation. Currently, this option is unselected as default, but the next release of VarSeq will have this option applied by default. Additionally, we will take multiple variants at a single position and split them, so each is represented individually. These two steps are important for optimal annotation of each variant separately and with consistent representations in the VarSeq variant table and GenomeBrowse.
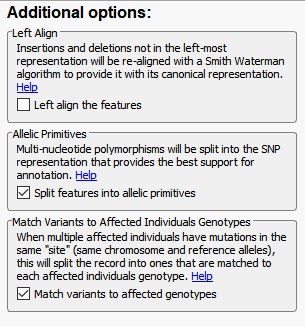
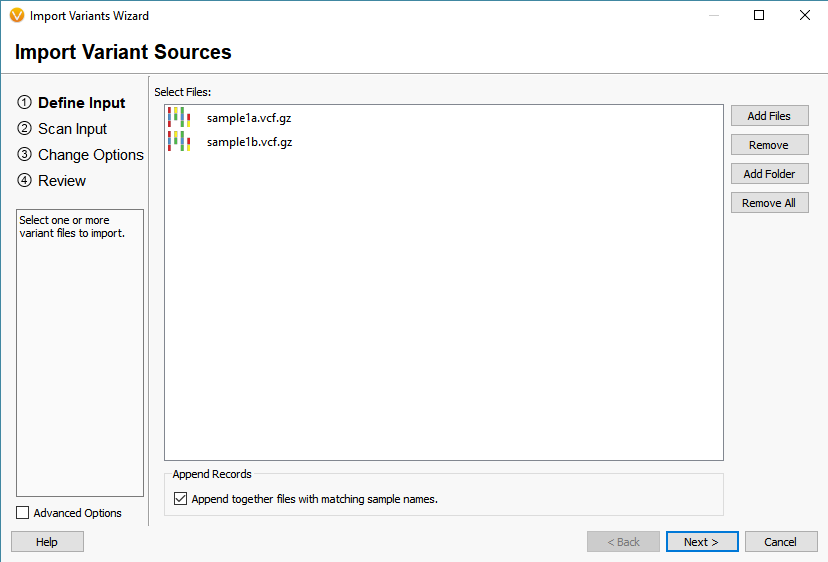
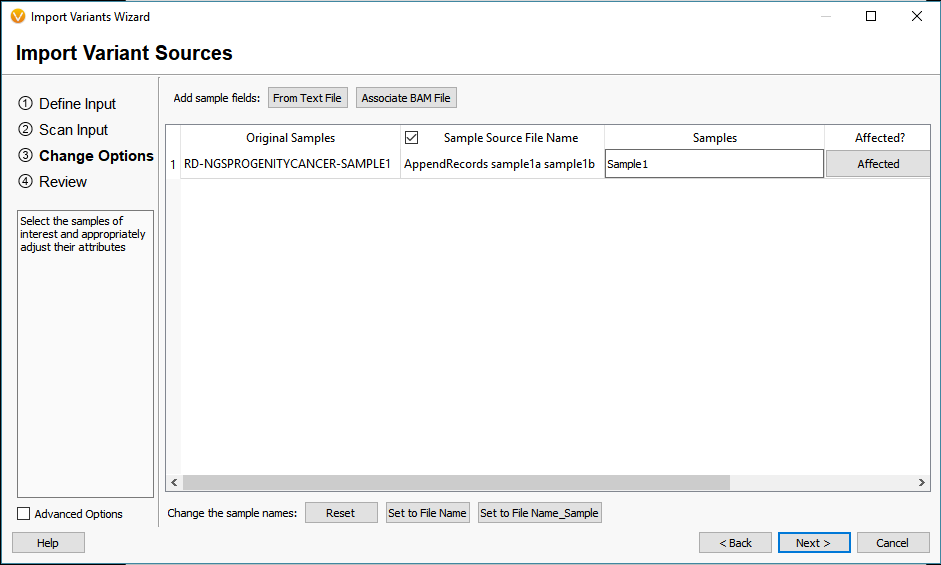
The last of the Additional options is to Match Variants to Affected Individuals Genotypes. Essentially, if the VCF contains multiple samples, this feature will ensure that any shared variant will be accurately matched to the GT (sample’s genotype) when imported into VarSeq. Another helpful tool for dealing with multiple VCFs is to Append together files with matching sample names (Figure 6). Whether you want to merge VCFs that represent individual chromosomes or merge indel and SNV VCFs, this tool will create a single representation of each of samples collection of variants across all VCFs.
The take-home message for this blog is that Golden Helix is aware of the intricacies of VCF format and how much VCF variety exists. We sought to create a universal import tool which minimally imports all fields in the VCF, but also gives the user the ability to modify the fields for an ideal workflow. If you have any specific questions regarding your data or how to build a VarSeq workflow, please contact us at [email protected] so we can assist you.



