Thank you to everyone who joined me for yesterday’s webcast, Using VarSeq Templates to Advance and Customize Variant Analysis, I hope you all enjoyed it. If you missed the live event and are interested in knowing what we talked about, good news, you can watch the recorded version right here!
There were so many great questions asked during our Live Q&A that I was unable to answer all of them, unfortunately. So, I thought it might be helpful to compile some of the Frequently Asked Questions for everyone!
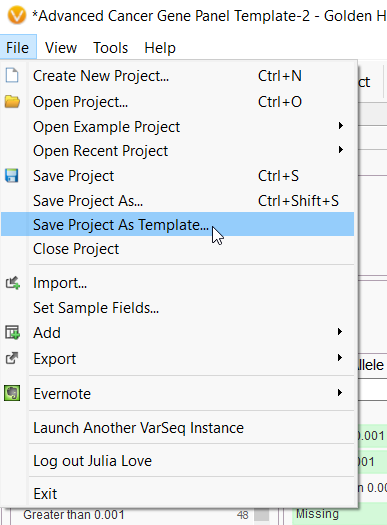
Q. How can I save my own project as a template?
A. Once you create a project, load the annotations/algorithms, and build the filtering logic you can go to the file menu and then save project as template. You then name the template and the next time you have new samples just select that template from the list and you will be ready to go!
Q. How can I visualize my variants with GenomeBrowse?
A. To open up Genomebrowse, click on the + next to the open tabs in the variant table and select Genomebrowse. To plot the variants in your sample, BAM file, or other annotation sources like ClinVar, use the plot icon in the top left-hand corner of the VarSeq interface.

When you click on a variant in the variant table, Genombrowse will automatically pull up the variant.
Q. I am working with GrCh38 data and I am wondering if there are templates available also in GRCh38?
A. Yes, all of the shipped templates are available for GRCH38. When you go to create a new project, just make sure to select GRCH38 from the dropdown.
Q. When an annotation source is updated, does the new version automatically get incorporated into the template?
A. The templates do not automatically incorporate newer versions of annotation sources. However, a new feature of VarSeq allows you to change these settings when you design the template. When you save a project as a template, select the annotations tab.
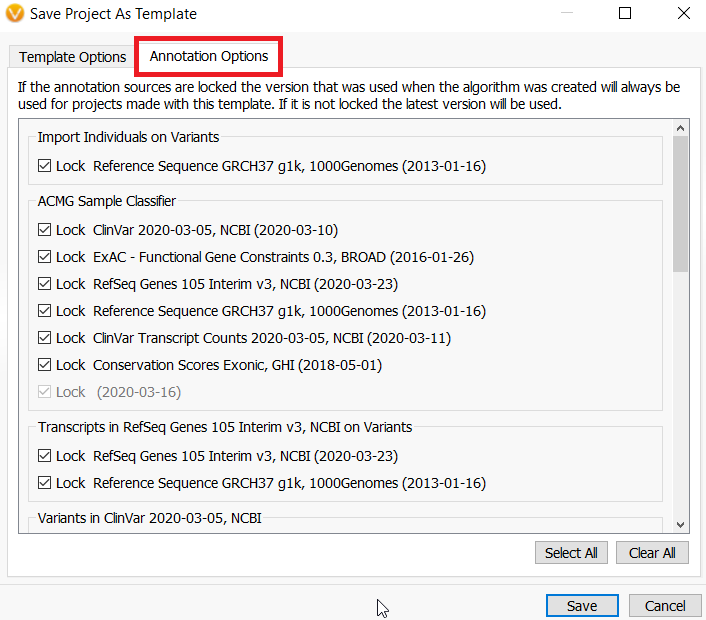
Everything is automatically set to locked. However, if you want to deselect any of these annotations, the next time the annotation is updated all projects made with this template would have the new version incorporated.
Q. Do you have any automated features for variant analysis that can use the project templates?
A. Yes, this can be done using VSPipeline. If you have a large batch samples you can use your template and automate the import and analysis process. Also you can export the resulting variants after filtering for reporting, or go into that project for continued evaluation with VSclinical ACMG or AMP.
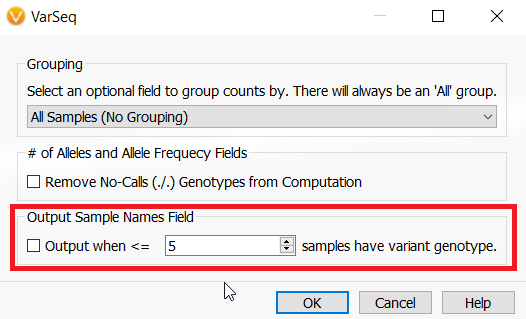
Q. Does the Count Alleles algorithm search for variants across all previous projects?
A. On a project level, no. Count alleles will search through samples and variants only within the project. However, if you are using VSWarehouse the count alleles algorithm is integral. You can use count alleles to search across samples and projects and as an annotation to prioritize variants that have low allele frequencies across all projects uploaded to VSWarehouse.
Q. Is there going to be an update to gnomAD frequencies in order to filter by ethnic groups?
A. For GnomAD, the fields are available in VarSeq to compute an allele frequency for ethnic groups/subpopulations, however, an alternate allele frequency field itself is not. Also, the allele frequency for subpopulations are included in VSClinical so they will be incorporated as part of the ACMG/AMP guidelines. However, we do have a version of gnomAD curated that has the allele frequencies for subpopulations calculated and it can be used in SVS as well. If you would like access to this annotation, please contact support at [email protected].
Q. How can you see which sample is carrying the variant?
A. The sample that you are currently analyzing is always shown at the top of the VarSeq interface and the sample name is displayed in the variant table.
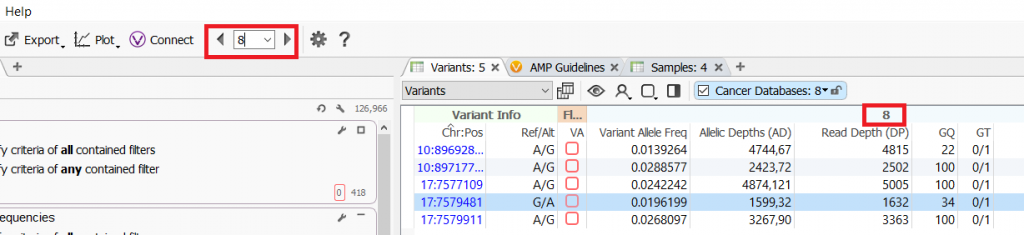
If you are wondering in which samples a variant is in common from the count alleles algorithm, there is an optional output from the algorithm to include sample names. If selected the Sample Names field will be in the variant table under the Count Allele algorithm output.

Q. Has the variant calling software used here been benchmarked against GATK?
A.Our partnership with Sentieon is a GATK based but dramatically improved variant caller. Essentially Sentieon uses more efficient computing algorithms and an enterprise strength software implementation. Combined, Sentieon DNAseq has no run-to-run differences because there is no down-sampling in the high coverage regions and has no thread dependency. With these improvements, Sentieon DNAseq won the precisionFDA Consistency. Truth. And Hidden Treasure Warm-up challenges and achieves higher accuracy than the original BWA-GATK pipeline by eliminating the randomness and noise from the software implementation while using the same mathematics. If you want to learn more about Senition you can check out their product page: https://www.sentieon.com/products/
Also if you want to reach out to support, we can schedule a demo/training session and see about setting you up with a trial license for Sentieon.
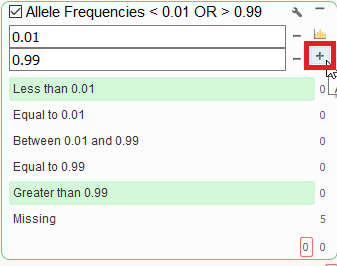
Q. Some reference alleles are extremely rare and we may not wish to ignore these when searching for mutations for rare disease. Not sure how to filter so these are included in VarSeq. MAF > 0.99?
A. You can create a filter that captures the top end .99% or higher or the 0.01% or less for the altering major, minor allele. To do this, first click on the small wrench icon the filter card, and then click on the small plus icon to add a comparison.
Q. In hardware prospects for this software. What IT specs are required?
A. The system requirements for VarSeq can vary depending on data size, server configurations, and proxy settings. I have provided the link for our VarSeq system requirements page which will provide the IT details for each scenario. However, there are separate system requirements if for VSWarehouse if you are planning on using VarSeq with VSWarehouse.
https://www.goldenhelix.com/resources/system_requirements.html
https://www.goldenhelix.com/products/VarSeq/VarSeq_Warehouse.html
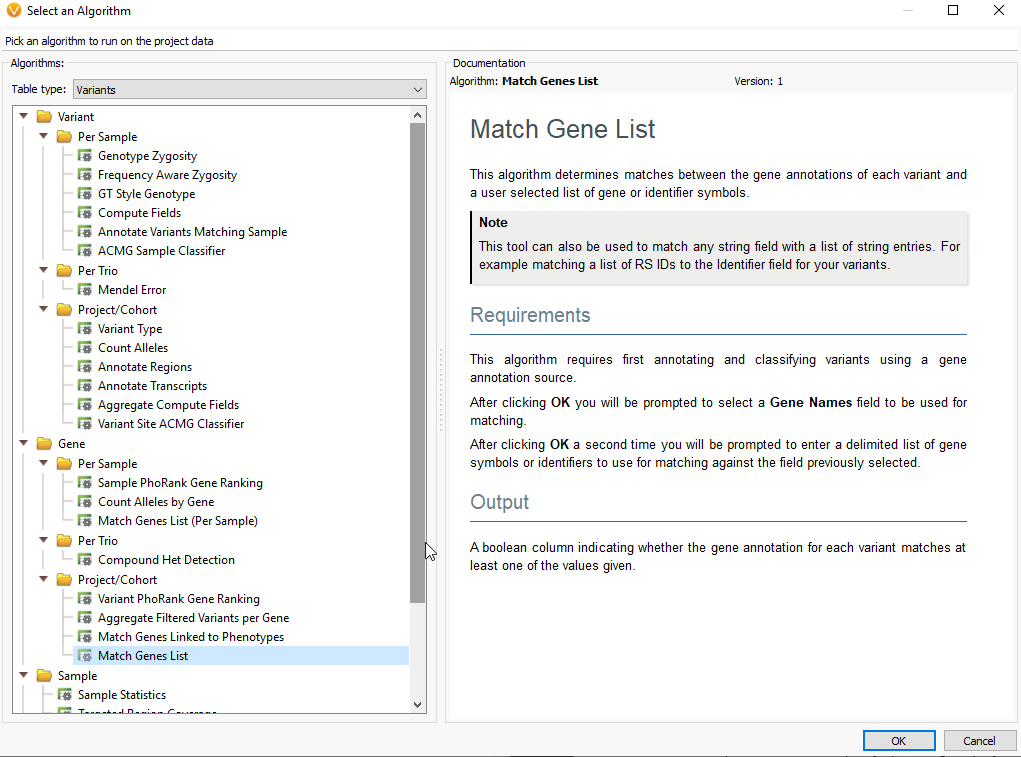
Q. How can we provide our own GOI?
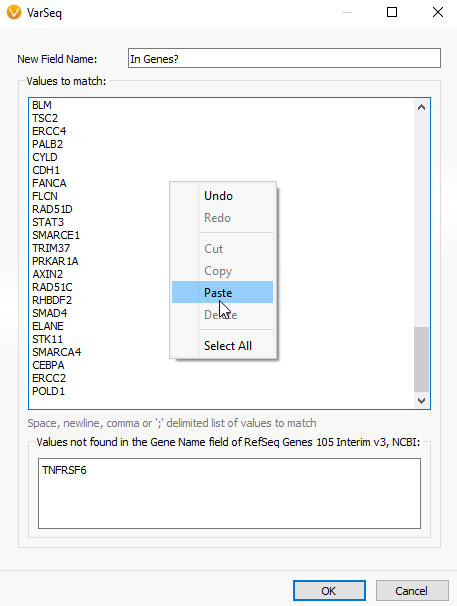
A. If you have a gene list perhaps as a word document, excel file, or text file, you can simply copy and paste that gene list into the Match Gene List window. To access the Match Gene List Algorithm, click on the Add Iconà Computed dataà Match Genes List.
Then just copy and paste the gene list into the “Values to match window”.
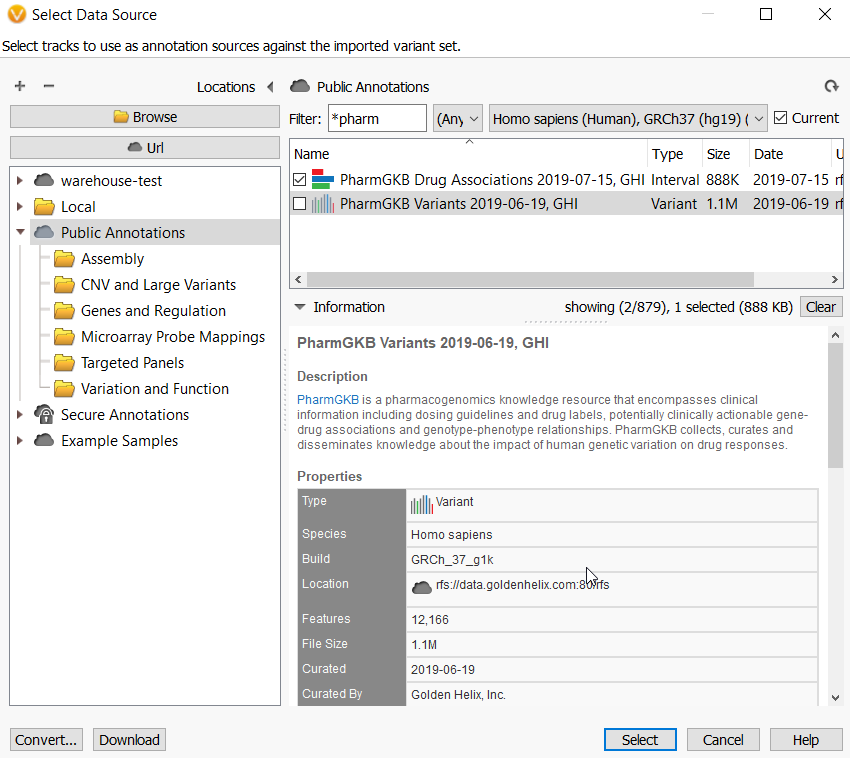
Q. Is this a good idea to go through ACMG annotated variants for checking a novel Pharmacogenomics identified variant? Or the functional prediction based on ACMG guideline would be useless for PGx phenotype prediction?
A. Since PGx variants are generally not within genes, they are somewhat predictive by simply being common SNPs shown by GWAS studies to be associated with different drug responses in patients. Thus, I would recommend using the PharmGKB annotation source which has clinical information abut drug labels, clinically actionable gene-drug association, and genotype-phenotype relationships. You can use the ACMG Classifier to point out gene modifying variants, but it has not prediction about what the variant means in terms of impact in drug metabolism or any other functional aspect.
I hope you found this to be useful in learning more about the VarSeq project templates. If you are interested in seeing VarSeq in a more personalized demo for your organization, please reach out to our team and we will be more than happy to schedule a call with you. Or, if you have any additional questions not covered here, please enter them down below!



