The ACMG classification guidelines for variant pathogenicity are as ubiquitous as they are complicated to implement. They play a consistent and evolving role in the standard workflows of many experts in the next-generation sequencing field, both in the clinical and research space. Furthermore, they can be effectively applied in both somatic and germline workflows. Hence, consistent and auditable methods for exploring the ACMG classification of variants are essential for saving time and maintaining accuracy in a high-throughput setting. In this blog, we will cover some of the fundamentals of the ACMG auto classifier and explore an important detail: under which circumstances to apply the Variant Site ACMG Classifier algorithm versus the ACMG Sample Classifier algorithm. Some readers may be pleasantly surprised to learn they can save some time and effort by being more discerning.
Many of our users are familiar with the versatility and intuitiveness of our automatic ACMG classification algorithm. For readers who are just getting started or are looking for more information, allow us to provide some general details before elucidating some of the nuances of applying the classification algorithm under different circumstances.
From a workflow perspective, there are two complementary ways to leverage the ACMG classification guidelines. The first is in a largely automated fashion to filter out benign and likely benign variants. The second is in a more granular setting to generate a clinical interpretation. The intuitiveness of this two-pronged approach is a major benefit of VarSeq’s capabilities; users can configure an accurate but not overly-specific workflow that identifies candidate variants for in-depth analysis. Hence, users never let go of the rigor and specificity required for accurate and reproducible classification.
Let’s consider the aforementioned workflow in a little more detail. The VSClinical ACMG or AMP module (Figure 1) encapsulates the rigorous, detailed, and exhaustive variant classification and interpretation step, ensuring that our users have the final say on a variant’s fate. In particular, users can perform a detailed analysis of each criterion and render a cumulative interpretation based on the information curated by our ACMG classification algorithm.
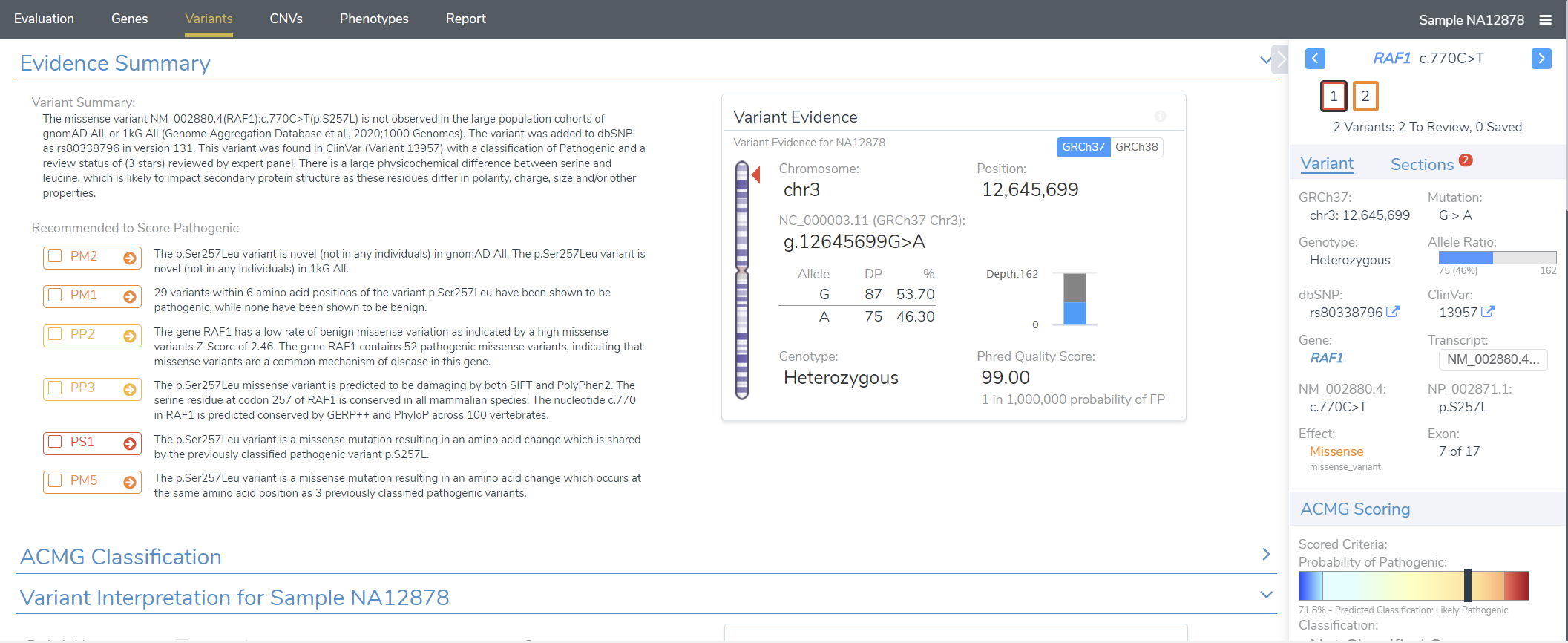
However, the real NGS workhorse is the filtration step. VarSeq has a completely open-ended approach to filtration, allowing users to integrate sources of their choice and use them in a robust filter chain (Figure 2). Our ACMG classifier is a popular choice as a filter and can be used quite variably. In the strictest of cases, users can use the ACMG auto classifier to narrow their search down to only pathogenic and likely pathogenic variants. This usually comes up in larger (e.g., whole exome or whole genome sequencing) germline projects. Conversely, users may want to be a bit broader and simply filter out obviously benign variants. This is often more expedient in somatic use cases or in smaller projects where users can afford to be more open-minded.
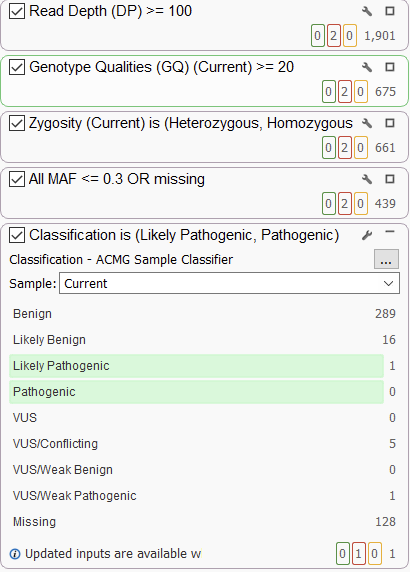
While there is clearly variability in how users can implement the classifier in a filtration strategy, the nuance extends beyond this application to the algorithm itself. Familiar users will note that there are two versions of the ACMG classification algorithm with slightly different use cases (Figure 3). Let’s take a minute to go over them and hopefully save some time for those currently developing workflows or reassessing existing workflows.

In short, the ACMG Sample Classifier analyzes each variant present in a project on a per-sample basis, whereas the Variant Site ACMG Classifier does not consider sample-level information. The most obvious practical effect of this is that, on average, and especially for projects with larger numbers of samples, the ACMG Sample Classifier will take longer to run. So when should users trade the longer run time for additional information?
We already touched on some of the filtration strategies that involve the ACMG auto classifier, which may shed some light on how we might suggest one version of the algorithm over the other. There are a few specific use cases that are best approached by running the ACMG Sample Classifier. In general, any time you want to consider the variant’s pathogenicity with respect to the patient, you probably don’t want to let go of the information included in the ACMG Sample Classifier. The main field that is considered here is zygosity, which is compared against the inheritance model for the variant to determine pathogenicity in the context of the patient.
An example of a use case where this is important is in trio workflows or hereditary disease investigations in general. Specifically, the ACMG Sample Classifier will score PS2 for de Novo variants. Furthermore, this may be broadly useful in single-sample workflows, but especially whole exome or whole genome situations, where we usually want to be stricter in our approach to narrow things down to a workable number of variants.
Conversely, in somatic use cases, we’re generally applying the ACMG auto classifier in our filter chain to filter out benign variants, as opposed to identifying pathogenic variants. Hence, in this case, we can afford to consider variants independently of their relationship to the sample. We, therefore, usually recommend running the Variant Site ACMG Classifier for somatic workflows.
Beyond the dichotomy between somatic and germline workflows, it’s often expedient to use the Variant Site algorithm over the Sample Classifier in cohort studies. This is for two reasons. First and foremost, as the sample volume in a project increases, the Sample Classifier algorithm will trend toward an exponential rather than linear runtime. Secondly, in cohort studies, we can afford to be a little more agnostic with regard to sample information since the end goal of our analysis is typically not a clinical report but information on variants present across the cohort.
To summarize, the main takeaway is that the ACMG Sample Classifier is more specific but slower, whereas the Variant Site ACMG Classifier is faster and agnostic to sample-level information, in particular zygosity. There are pros and cons to both, and we hope that the details provided in this blog have provided some clarity and possibly improved the throughput of some of our users.
While these recommendations are a great place to start, consider this blog post as a foot in the door more so than a list of hard and fast rules. Hopefully, after reading this, users will be comfortable with the differences between the algorithms.
Golden Helix has developed innovative tools for the clinical interpretation of variants based on the ACMG guidelines. The guided workflow enables following the ACMG guidelines used to identify and classify causal variants for inherited disease risk, cancer predisposition, and the diagnosis of rare diseases. This integration drives deeper analysis and accelerated interpretation with the benefit of manual inspection control.




