In February 2020, the American College of Medical Genetics (ACMG) and the Clinical Genome Resource (ClinGen) published a joint consensus on standards for the interpretation and reporting of copy number variants (CNVs) ranging from large CNVs spanning multiple genes to small intragenic events1. The guidelines consist of over 80 different criteria which are arranged into five distinct sections. These extensive guidelines have proven challenging to configure and implement in a clinical setting. Golden Helix is the first commercial company to incorporate these guidelines into a user-friendly product for CNV interpretation and clinical reporting2. The guidelines are integrated seamlessly into our VSClinical variant interpretation hub, enabling automated CNVs analysis with the same ACMG based five-tier classification system we have used for small variants. This blog will review how our product automates CNV scoring according to the ACMG/ClinGen guidelines and provides the user a step-by-step guide through the five sections to arrive at a final CNV classification.
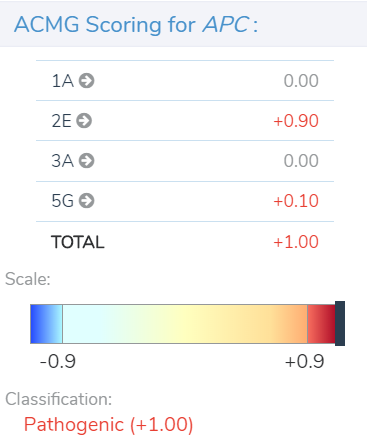
After a user imports and filters CNVs in VarSeq, they can access the tool for CNV clinical analysis and reporting in VSClinical. Within the VSClinical CNV tab, each of the five sections presents the user with an automated score and/or a carefully crafted series of questions about the CNV, the patient, or relevant literature to simplify the assessment of specific ACMG criteria. As each section is scored, points are accrued or subtracted to arrive at a final pathogenicity score, ranging from≤ -0.99 for benign to ≥0.99 for pathogenic, as shown below in our example of a pathogenic heterozygous deletion in the adenomatous polyposis coli (APC) gene.
Section 1 is a first-pass assessment that determines if the CNV affects a protein-coding region or other known functionally important region. If that effect is confirmed, a score of 0 is assigned and the evaluation can continue, whereas, if no such effect is confirmed, the evaluation ends here.
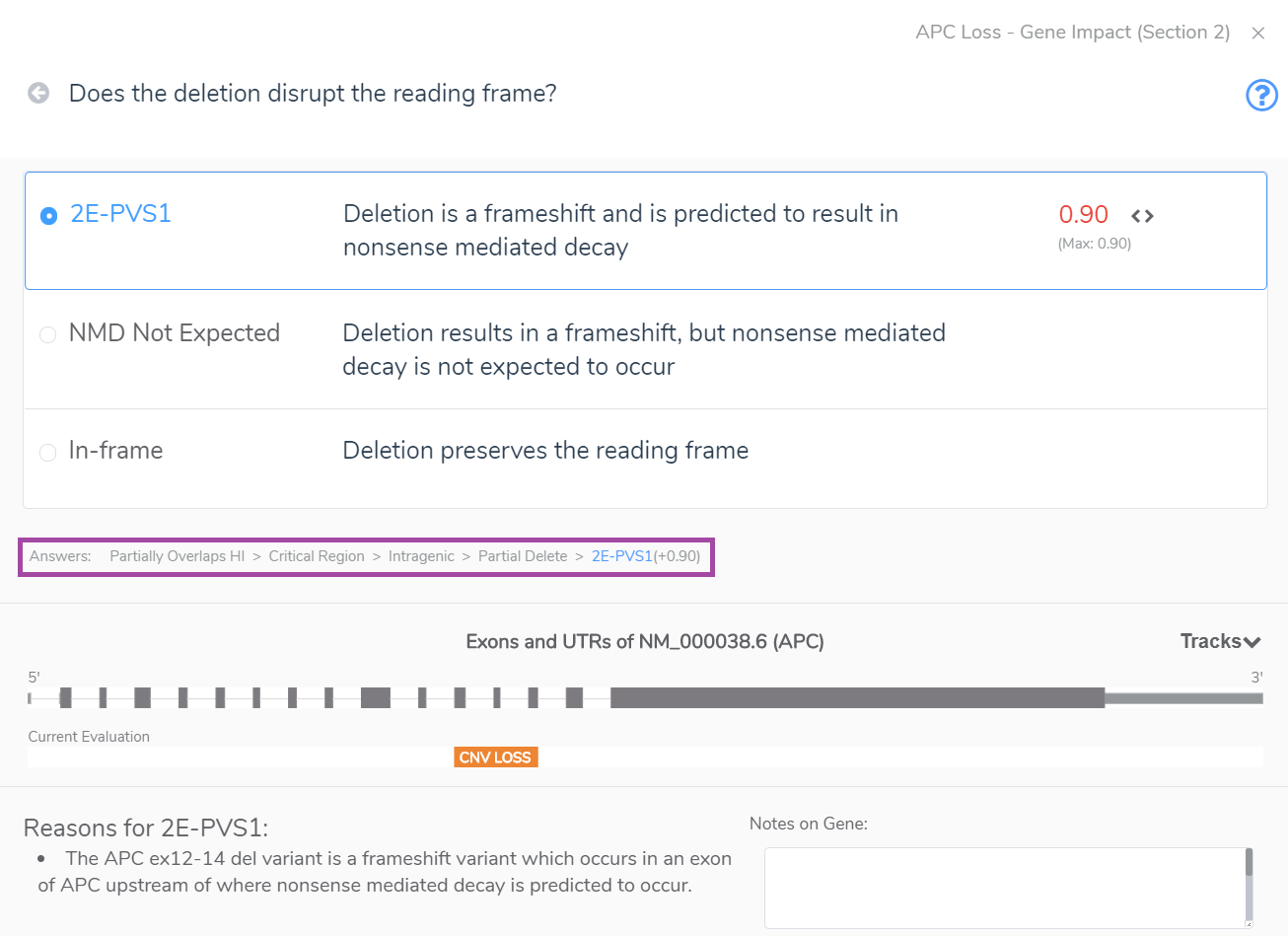
Section 2 unearths whether the CNV overlaps with gene regions that have established dosage sensitivity, i.e., does it cover a gene(s) with known haploinsufficiency or triplosensitivity? The example below shows an intragenic CNV which spans only three exons of APC, a gene with a known record of haploinsufficiency in the ClinGen Dosage Sensitivity Map. Notably, such evidence can also come from the user’s internal clinical database. CNVs that span a gene(s) with known dosage sensitivity can be immediately classified as pathogenic, but partial overlaps as seen below require additional evaluation to arrive at a final classification. In this case, the CNV accumulates a score of 0.9 at the end of Section 2 because this partial deletion of a haploinsufficient region cases a frameshift that results in nonsense-mediated decay. The answers to the Section 2 series of questions are highlighted in the rectangle below our multiple-choice menu.
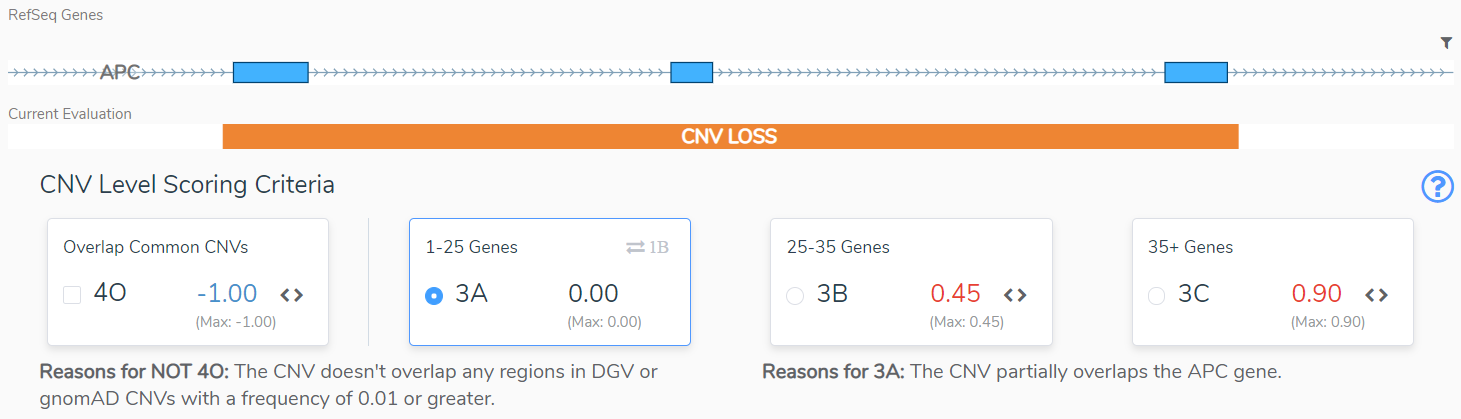
Sections 3 and 4 take us through size considerations and the genomic context for the CNV. In Section 3 we typically consider size as an indication of the severity of a CNV. The larger the CNV, the more pathogenic it is scored to be, but there are notable exceptions, such as when a large cytogenetic event only overlaps benign regions2. If a CNV does not overlap a region with known dosage sensitivity, in Section 4 we are asked to evaluate the clinical significance of the overlapping regions. This will be based on Population Evidence, Case-Control Studies or Individual Case Evidence gleaned from the literature, public databases, or internal clinical lab data2. This APC CNV (Figure 3) does not overlap any regions that are benign or variants that are common in the healthy population, and as such does not lose any points in Section 4, but also does not gain any points in section 3. Based on its small size, 3A is selected and no points are added.
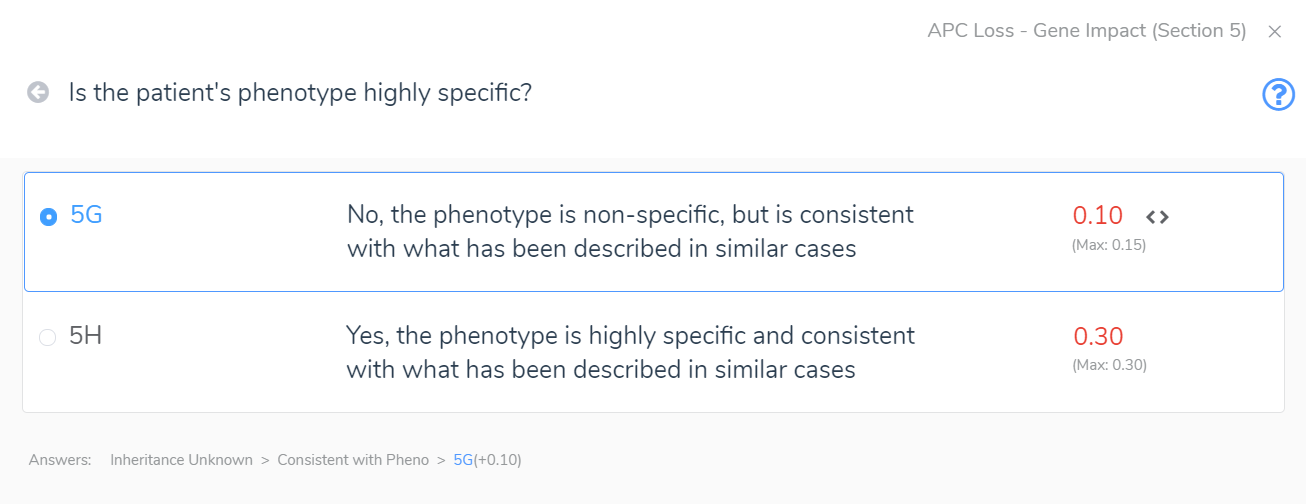
Section 5 adds the final layers of evidence. In this section, we are brought through a series of questions to evaluate the inheritance pattern of the patient’s CNV and determine how specific and consistent their phenotype is with what is expected for the CNV event. Even with an unknown inheritance and a non-specific but consistent phenotype, in this example (Figure 4) we accumulate an additional score of 0.1. This brings us to a final pathogenic score of 1 for the heterozygous deletion in APC.
In conclusion, the extensive CNV interpretation guidelines developed by the ACMG and ClinGen organizations can be understandably complex to implement in a clinical setting, but we have made it simple. Any clinical or research lab wanting to streamline their CNV analysis workflow while maintaining this high standard need look no further than VSClinical!
Thank you for reading this article! If you enjoyed this content, please check our other blog posts here! To learn more about CNV analysis in VSClinical and any of the other products in our VarSeq suite, please reach out to us at [email protected]. If you have any questions or concerns about the topic presented in this blog or about any of our software, please comment below or email us at [email protected].
References
- Riggs E, Andersen E, Cherry A, Kantarci S, Kearney H, Patel A, Raca G, Ritter D, South S, Thorland E, Pineda-Alvarez D, Aradhya S, Martin C (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med, 22(2), 245-257.
- Fortier N, Rudy G, & Scherer A. (2021). Implementing the ACMG Guidelines for CNV in a Commercial Software Solution. The Journal of Precision Medicine, 7(1), 43-49.



