
It may come as a surprise to our long-standing users that the multi-allelic import and representation in VarSeq is slated to have different import options with the 2.5.0 upgrade. As the software grows and evolves, we strive to meet the changing needs of our users, and this is one area where an update was sorely needed. Before going into an overview of the changes, we wanted our long-time users to know that any project templates created pre-2.5.0 will still retain their saved variant import options, so there is no need to worry this will change your current workflows. We wanted you to keep this information in mind when building new workflows in the future.
The Past
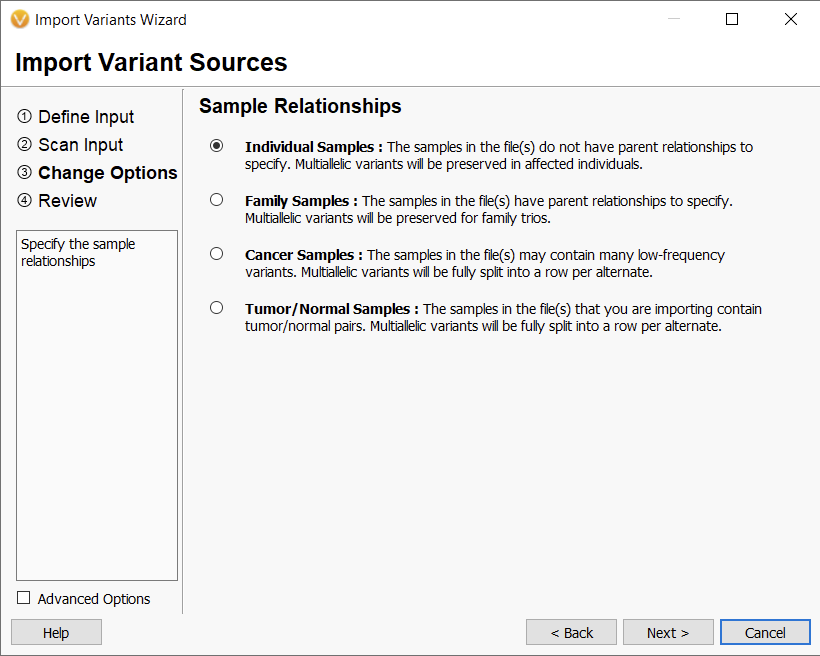
Previously, with version 2.4.0, we gave the option to change the Sample Relationships (Figure 1), which overall would designate a sample as germline or somatic. For the germline samples, multi-allelic variants would be preserved, and the idea was this could be useful when looking at inheritance patterns. For somatic samples, multi-allelic variants would be split into separate rows, which would be advantageous for annotating and filtering.
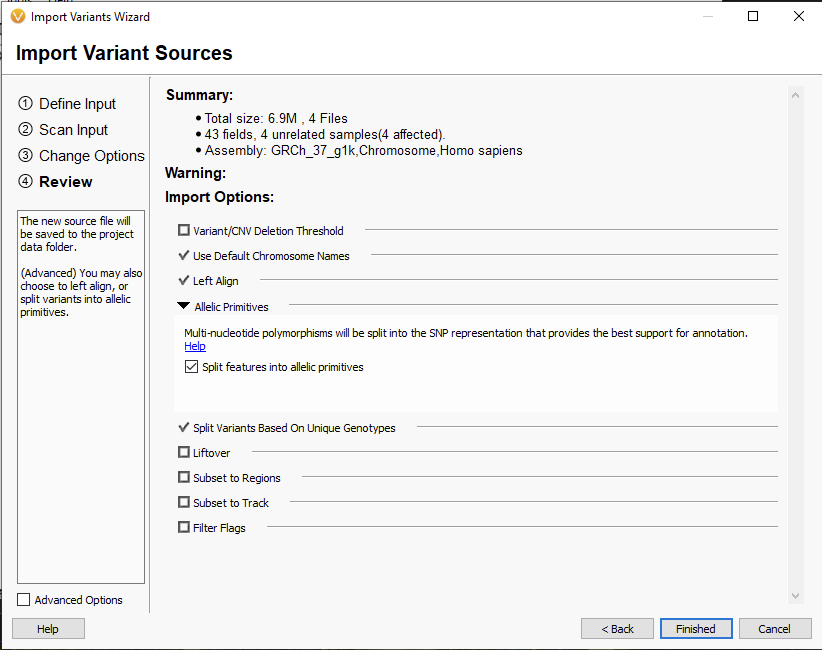
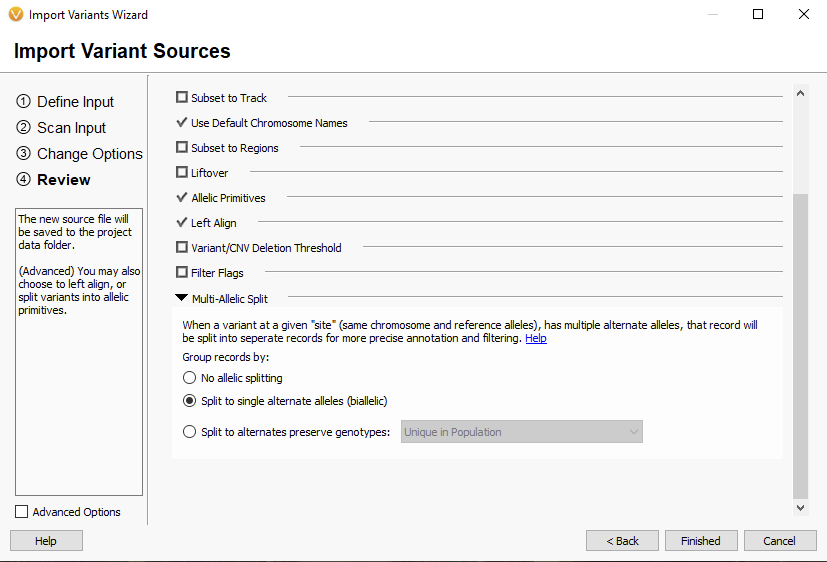
After that, we had the option of splitting allelic primitives into separate SNPs or leaving them intact (Figure 2). This option can be found on the ‘Review’ page of the Import Variant Sources. An example of an allelic primitive split could be an AG/CT change, which would then be represented as separate A/C and G/T calls.
The Present
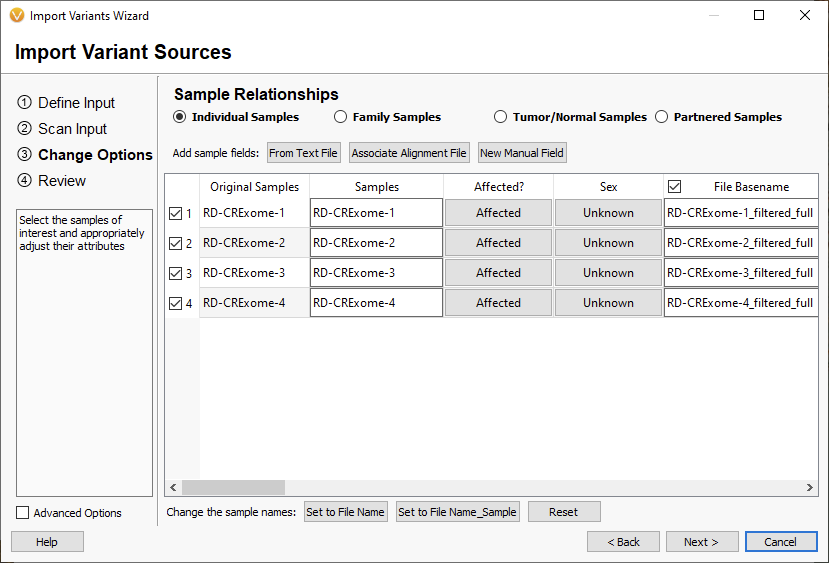
Now we are taking a look at the 2.5.0 import options (Figure 3). You will see there is no longer a ‘cancer’ designation that alters how the multi-allelic variants are represented. That is all handled on the Review page. With the Family Samples, Partnered Samples, or Tumor/Normal Samples options, you will be able to designate relationships between samples in the sample table. Otherwise, single and somatic samples will be treated the same during variant import. Switching between these options will change the default sample fields shown in the table but not how the variants are imported.
Moving on to the Review section (Figure 4), you can see that the Allelic Primitives Option is in the same place as before and has not changed.
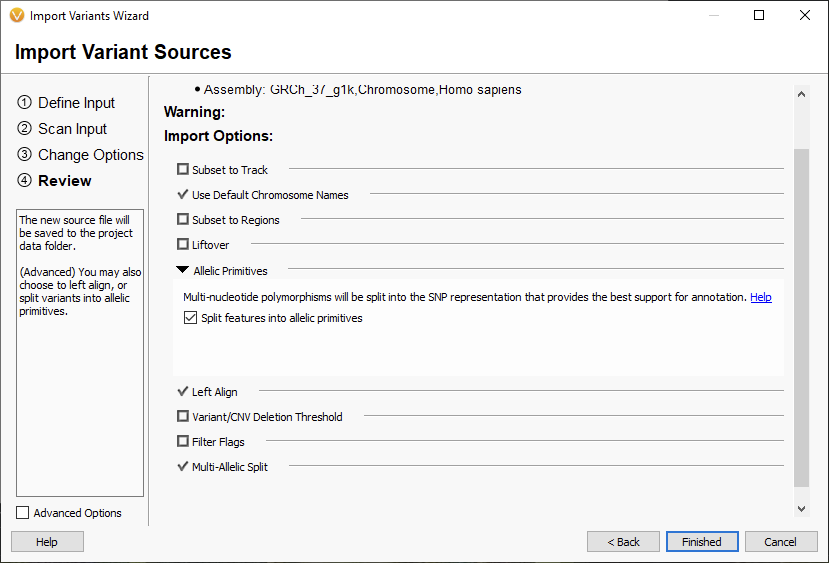
The big change to this section is the multi-allelic split, which previously was the big differentiator between germline and somatic samples. Now by default, this is applied to all sample types (Figure 5). Let’s break down what this option can do. The multi-allelic split is used when there are multiple alternative alleles at a single site. For example, the variant 3:37053568 A/G/T would be split into two separate records:
3:37053568 A/G
3:37053568 A/T
While the simple biallelic representation is appropriate for most workflows, there may be niche workflows where other representations may be more appropriate. Splitting the alleles provides more precise variant annotation and application of filters in the table view. However, alleles can be split to preserve genotypes to be unique in:
- Unique in Affected Family Members, which splits alleles into groups based on the affected sample (Affected individuals are not split apart). This may be helpful when looking at a family when you do not want to split apart genotypes to have two alternate alleles. Perhaps you have a specific compound het variant where the mother and father have different alleles. Note that this representation of alleles may compromise the annotation step, though, as many annotations are less precise when matching against multi-allelic calls.
- Unique in Population, which splits alleles into groups that represent each of the unique genotypes in the population. This could be helpful when lacking sample relationships, but when you want to keep the sample genotypes together, perhaps if there is a variant on both haplotypes. See the note in the above bullet point concerning annotations.
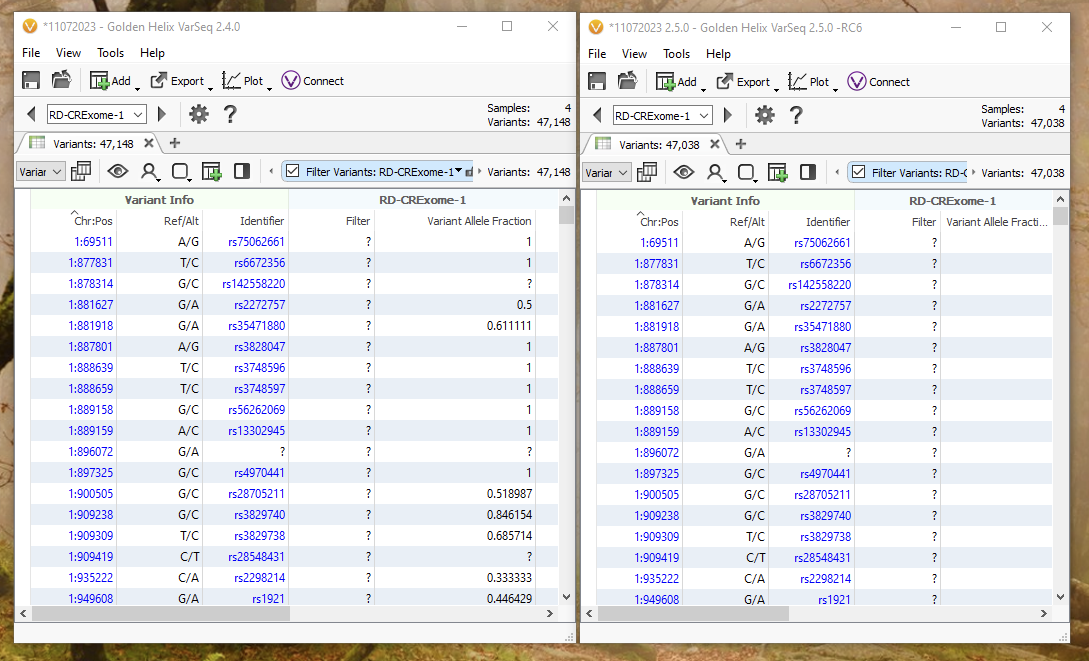
Multi-allelic splitting can be a bit hard to visualize, so let’s break down an example of how a complex variant is handled in 2.4.0 versus 2.5.0 with default import options for the same four germline samples. Version 2.4.0 can be found on the left, while version 2.5.0 can be found on the right (Figure 6). Although seeing a slightly different number of variants between the two versions may seem alarming, all differences can be tracked between how those complicated positions are being represented across multiple samples.
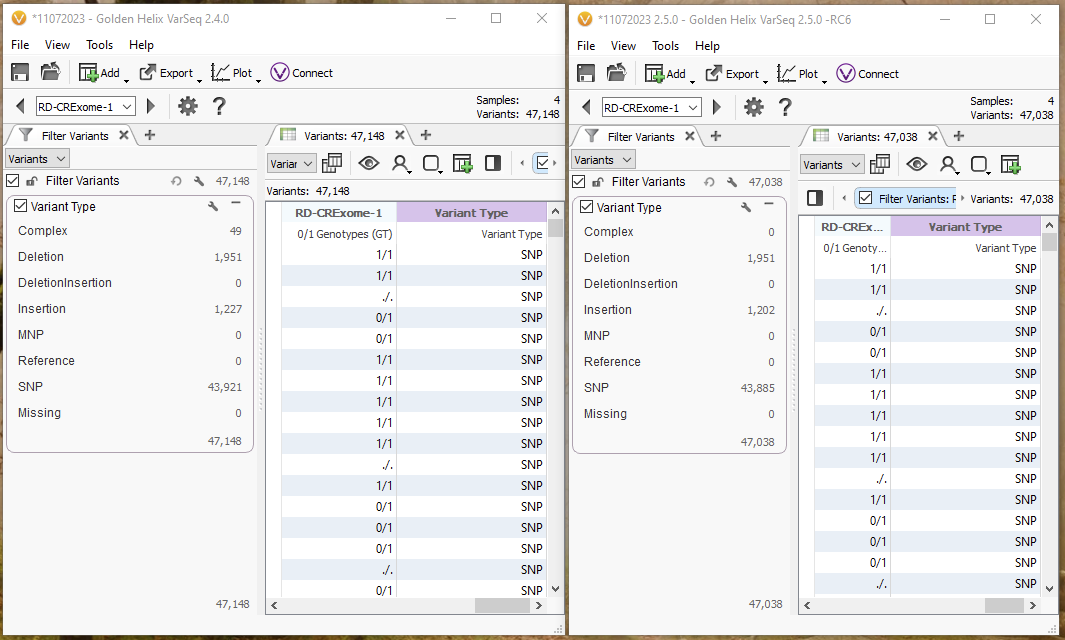
One way to look at this breakdown is to use the Variant Type Algorithm that can be found under computed data. Before, in 2.4.0, there were 49 variants that fall into the bucket of ‘Complex,’ while in 2.5.0, these are now shown as deletions, insertions, and other SNPs (Figure 7).
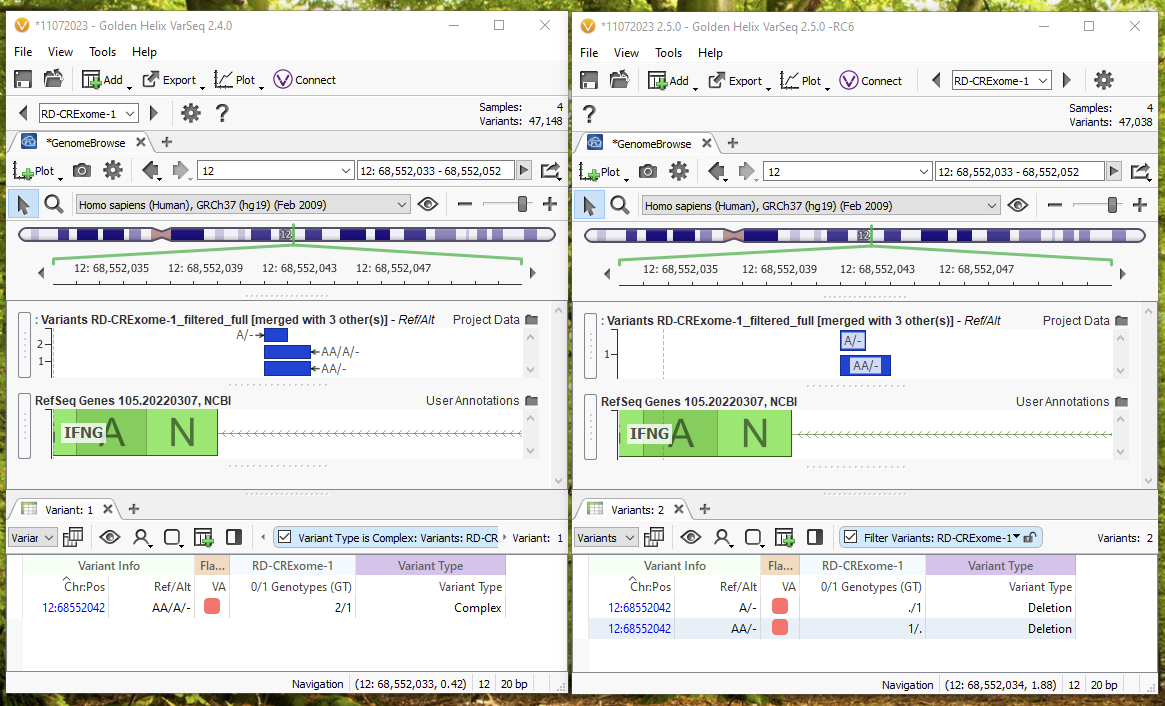
Let’s break down a specific example (Figure 8). Again, 2.4.0 is on the left, and 2.5.0 is on the right. For this multi-allelic call in Chromosome 12, 2.4.0 treats the call of AA/A/- as a complex variant with a 2/1 GT field. Meanwhile, 2.5.0 on the default import options will now be able to break this into two unique variants that can be individually annotated and filtered because of the multi-allelic split. In 2.5.0, we would see one variant as A/- with a ./1 GT field, and another as AA/- with a 1/. GT field. With the representation in 2.5.0, we will be able to separately filter and annotate each of the two calls, instead of considering them one overall variant.
While change can be uncomfortable, we believe this control over the display of complicated variants does clear up user problems seen with previous VarSeq versions. When moving into 2.5.0, make sure to always validate your workflows. For more information on this topic, please see this validation webcast.
We would also like our long-time users to remember that workflow templates made in previous versions will be maintained for the importing options with which they were originally created. As always, if you have any questions or concerns about your workflows, please contact support at [email protected].



