
As many of you may already know, we just released VarSeq version 2.5.0 this month! We have talked about the two headlining features a bunch, but we have not focused on what else has changed in VarSeq 2.5.0 that might also strike your fancy.
For those of you who might just now be tuning into the hype around 2.5.0, it is worth mentioning that those two main features of VarSeq 2.5.0 are:
1) The oncogenicity scoring engine from VSClinical can now be applied to the filtering and variant prioritization of VarSeq via the Cancer Classifier algorithm. We have a webcast that deep dives into the details of how this algorithm works and how you can include it in your workflow. Also, you can look forward to our upcoming webcast wherein we will apply the Cancer Classifier from a user’s perspective!
2) Introduced a carrier workflow and multi-sample analysis. The feature includes the new Couples Carrier Screening project template, a new paired partner import, the new Shared Carrier Gene Detection algorithm, and multi-sample analysis and reporting in VSClinical.
But now, let’s talk about a few lesser-known features of the new release that are worth mentioning.
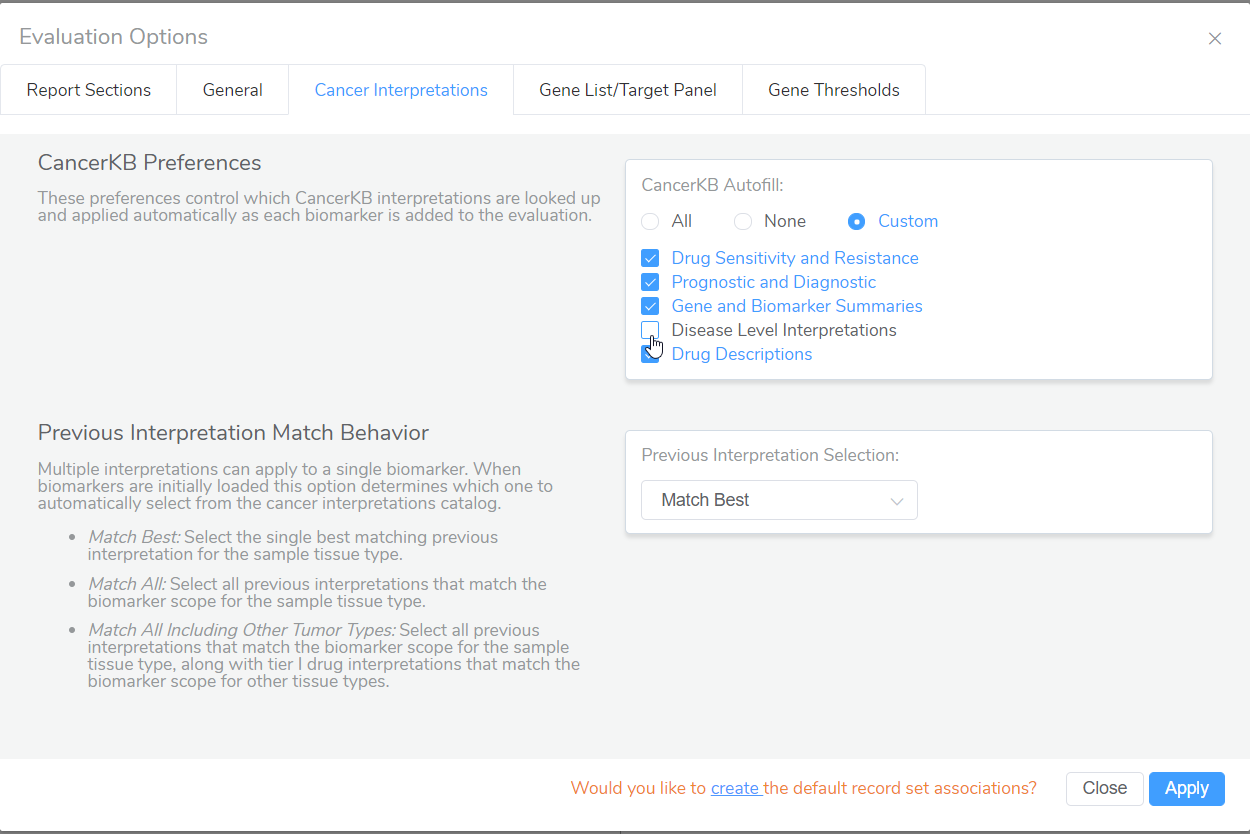
- Regarding Golden Helix CancerKB, a new Golden Helix CancerKB Genes source has been incorporated into VSClinical to fill in cancer gene details for the role in cancer, cancer hallmarks, somatic mutation types, and relevant tumor types. In addition, to give users more control over which interpretations from Golden Helix CancerKB will automatically populate into evaluations, a new Evaluation Option under Cancer Interpretations has been added to the AMP workflow.
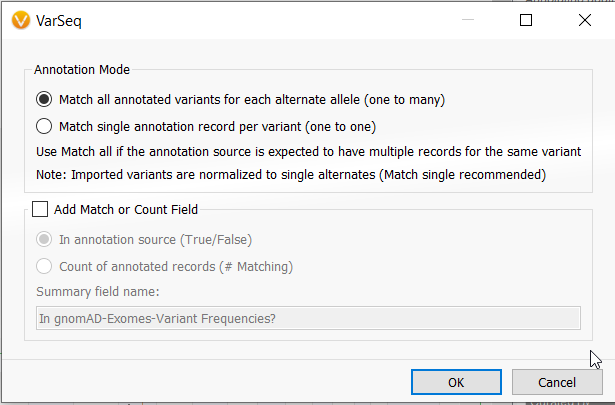
- The Variant Annotation algorithm used most commonly to annotate population catalogs, can now specify whether to use a “match one” or “match all” mode. It will also normalize multi-allelic variants to annotate the individual alleles that have different genomic coordinates than the table record. Variants with a reference for the alternate allele will no longer annotate against non-reference variants.
- The transcript modeling algorithm has been updated to handle the mitochondrial genes that rely on polyadenylation to complete the UAA stop codon. These previously were annotated as “stop codon absent” and did not have their HGVS pDot annotated, but they will now have the missing ‘A’s added to complete the transcript and provide protein impact transcript annotations.
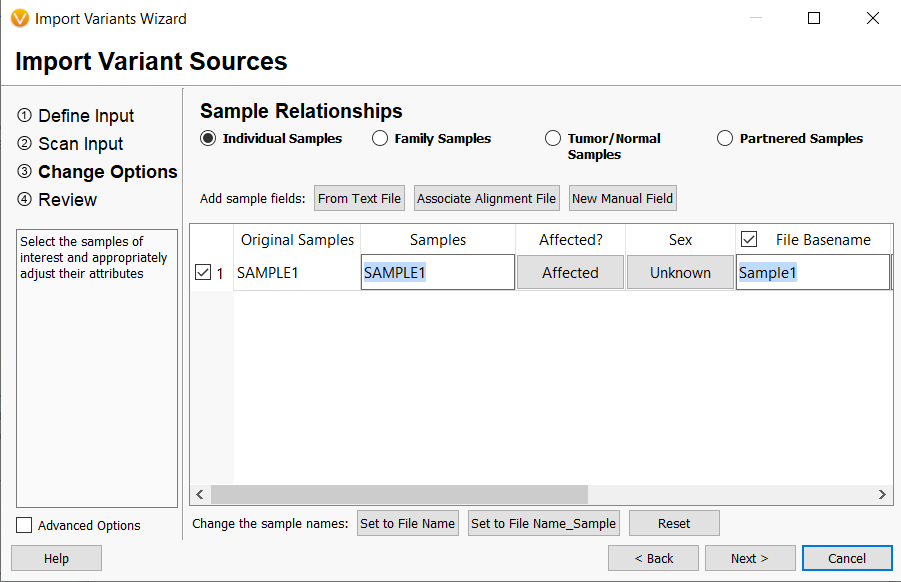
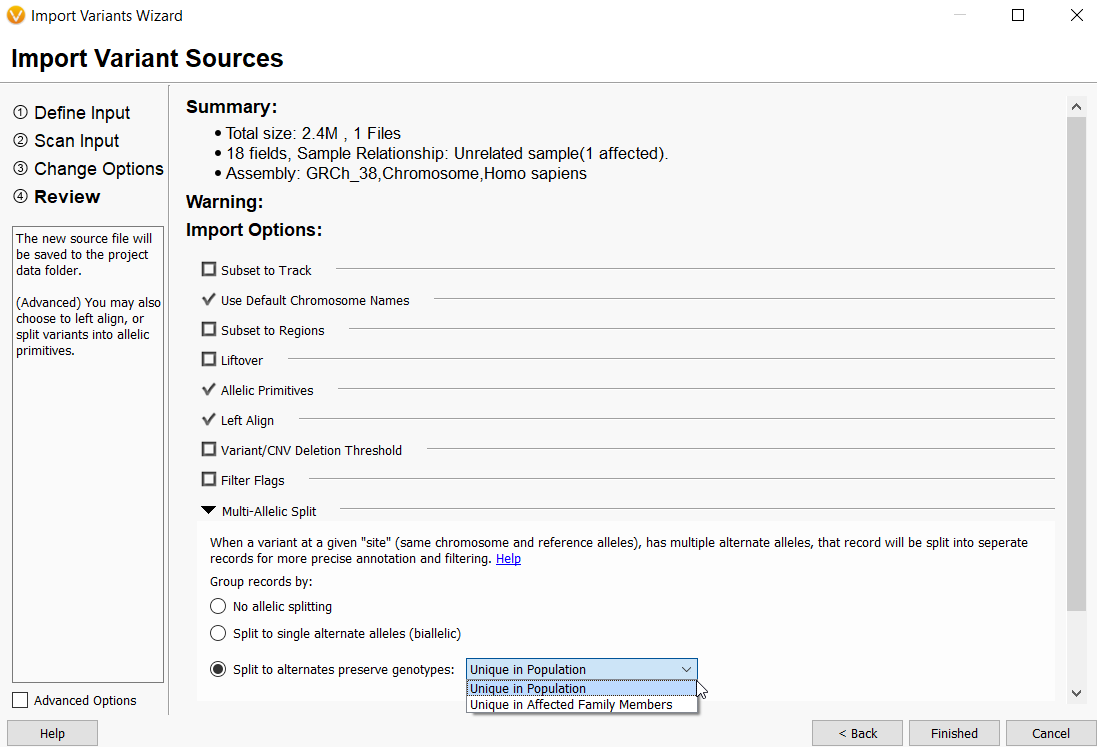
- The VarSeq importer has been updated to allow more flexibility and control over building project templates. This was achieved by separating the selection of sample relationship from multi-allelic splitting behavior. Previously, sample relationships were paired with multi-allelic splitting behavior and could not be altered independently by the user. The sample relationships are now 1) Individual Samples, 2) Family Samples, 3)Tumor/Normal, and 4)Partnered Samples. The multi-allelic splitting options are now 1)No allelic splitting, 2)Split to single alternate alleles (biallelic), and 3) Split to alternates and preserve genotypes either unique in population or unique in affected family members.
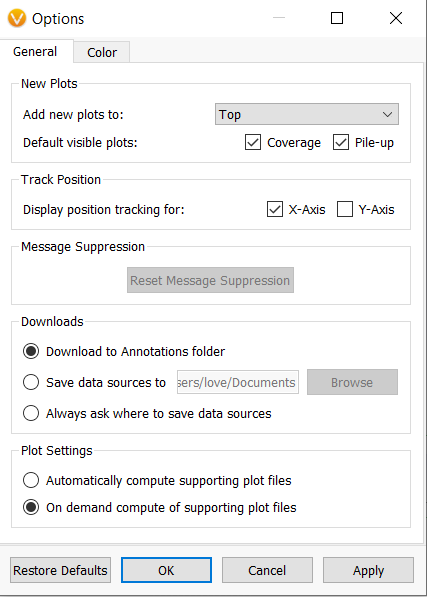
- In the GenomeBrowse options, you can now choose not to automatically compute supporting plot files such as the “.covtsf” created side-by-side BAM or CRAM files; in addition, the compute coverage task that runs on a BAM or CRAM plot that does not have a .covtsf could not be canceled. This has been updated, so clicking the “X” on the progress bar in the top-left stops the coverage computation and does not restart it.
I hope this blog has helped highlight some of the features that you can now access with VarSeq 2.5.0. I encourage you to review all of the changes within VarSeq 2.5.0 by browsing the release notes!



