Manually converting FASTQs to VCFs, importing these into VarSeq, and building projects from scratch is adequate when you have only a handful of cases per week. But as you start ramping up production, the key to your lab’s success quickly becomes how quickly and efficiently you can get to the reporting of your analysis. This blog will explain how you can automate the VCF and VarSeq Project generation process that requires only a few commands. This will expedite your path to analysis as the newly created project will be ready to import rare variants quickly into VSClinical.
For converting from FASTQ to BAM to VCF, I am using Sentieon. This is a secondary analysis tool from our partner company that provides the command line tools needed for generating variants.
Preparing Your Project Template
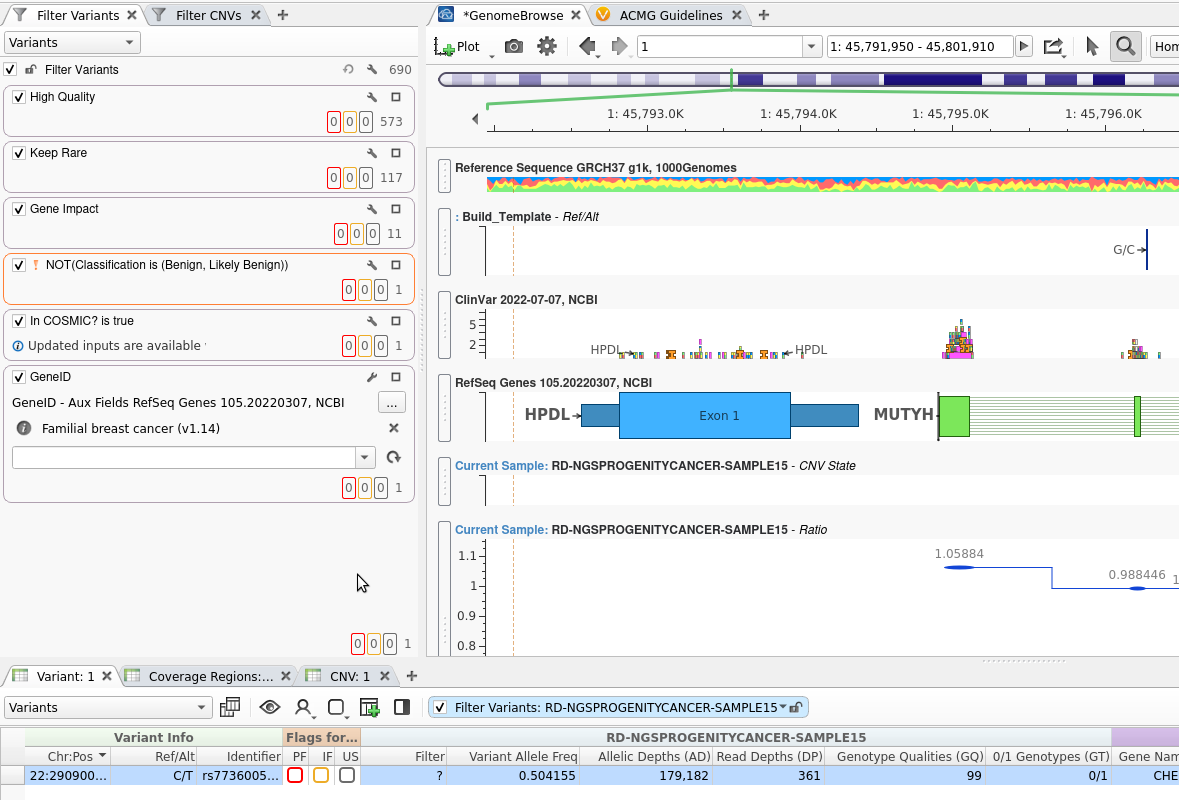
It is important to start your analysis pipeline from a well-built template. In Figure 1, I have a familial breast cancer workflow, and you can see that I have:
- Run coverage statistics
- Called my CNVs
- Have a strong filter chain present
These steps bring me down to my clinically relevant variant, and you can see that GenomeBrowse has already been pre-filled with the fields relevant to my analysis. Now, when I save this template, all of the instructions for running CNVs, filtering, and annotating will be carried with it! I will save this template as “Familial Breast Cancer with CNVs Workflow”.
If you want to know more about saving templates to protect workflows, please check out my recent blog, Locking Down Clinically Validated Workflows for Routine Analysis.
Alignment and Variant Calling with Sentieon
Now that the workflow template is ready to accept new VCFs, we are ready to move into Sentieon. In Figure 2, below, you can see two of my raw FASTQ files, as well as call_variants_pipeline.sh, which is my call-variants script. As you can see, I am doing this all in MobaXterm, which allows me to work inside of our server’s command line environment from my Windows computer. Sentieon can be run in a Linux environment, or this can be achieved from a Windows platform through Cygwin or a MobaXterm outlet.
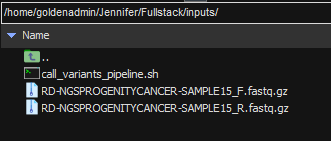
One directory up in our Linux server, I have a master folder with my various scripts, input files, a sample manifest, and my VSPipeline script (Figure 3). The sample manifest has been made before running Sentieon and VSPipeline, providing a text source from which VSPipeline can auto-fill patient and other relevant information.
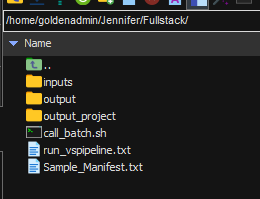
To get Sentieon started, I am going to input the locations for the following (Figure 4):
- the b
atch script - the c
all_variants_pipelinescript - the input VCFs
- the new output location
Sentieon will ask me to confirm these input and output directories before proceeding.
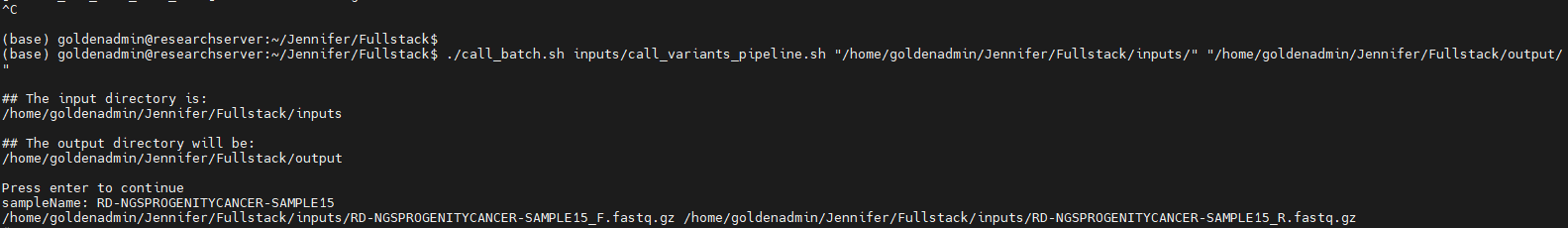
As Sentieon runs the alignment and variant calling steps, I can take a look at the call_variants_pipeline script that is feeding instructions to Sentieon (Figure 5). Some of these fields include the build for the VCF, the sequencing origin of the FASTQ, the input variables, and output sources.
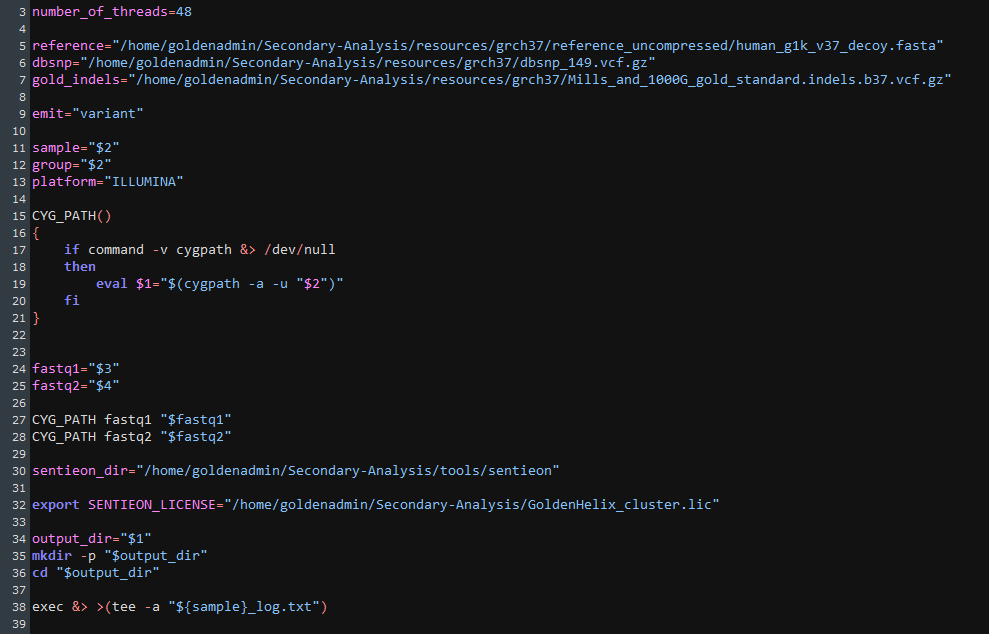
Sentieon will then work through the typical alignment steps, including mapping reads with BWA-MEM, deduping those reads, realigning INDELs, re-calibrating the final BAM, and generating the VCF in our preferred build.
After completing the VCF and BAM generation, this process can be automated further! The last line of Sentieon, in Figure 6, triggers the VSPipeline script to take over, funneling the VCF and BAM into the pre-made project template.

VSPipeline itself is the GUI-less VarSeq program for batching the creation of many projects at once. The run_vspipeline script here is quite simple, directing VSPipeline to create a new project with the template Familial_Breast_Cancer_with_CNVs_workflow created earlier (Figure 7).

Looking to bring repeatable clinical workflows to your lab?
Next, we can direct VSPipeline to the sample manifest and list of VCFs ready for import (Figure 8).

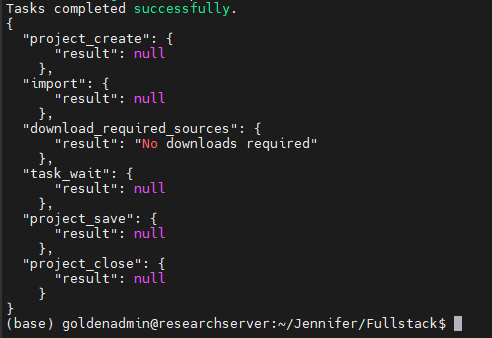
The last set of instructions tells VSPipeline that once the project is done rendering, it will save that project and close (Figure 9).

Meanwhile, it has only been several minutes, and our Familial Breast Cancer project has finished running in Sentieon and VSPipeline (Figure 10).
Ready for Final Analysis in VSClinical
Looking at the output project folder, I can see that the new project is ready and waiting, along with some familiar VarSeq files like data and project.log (Figure 11). When I launch VarSeq with the new project, I can easily inspect my work.

VarSeq, in GUI form, launches and brings up my Familial Breast Cancer project (Figure 12). At this point, all of the filtering is done, the CNVs are called, and the variants are ready to be imported into VSClinical for variant classification and final analysis.
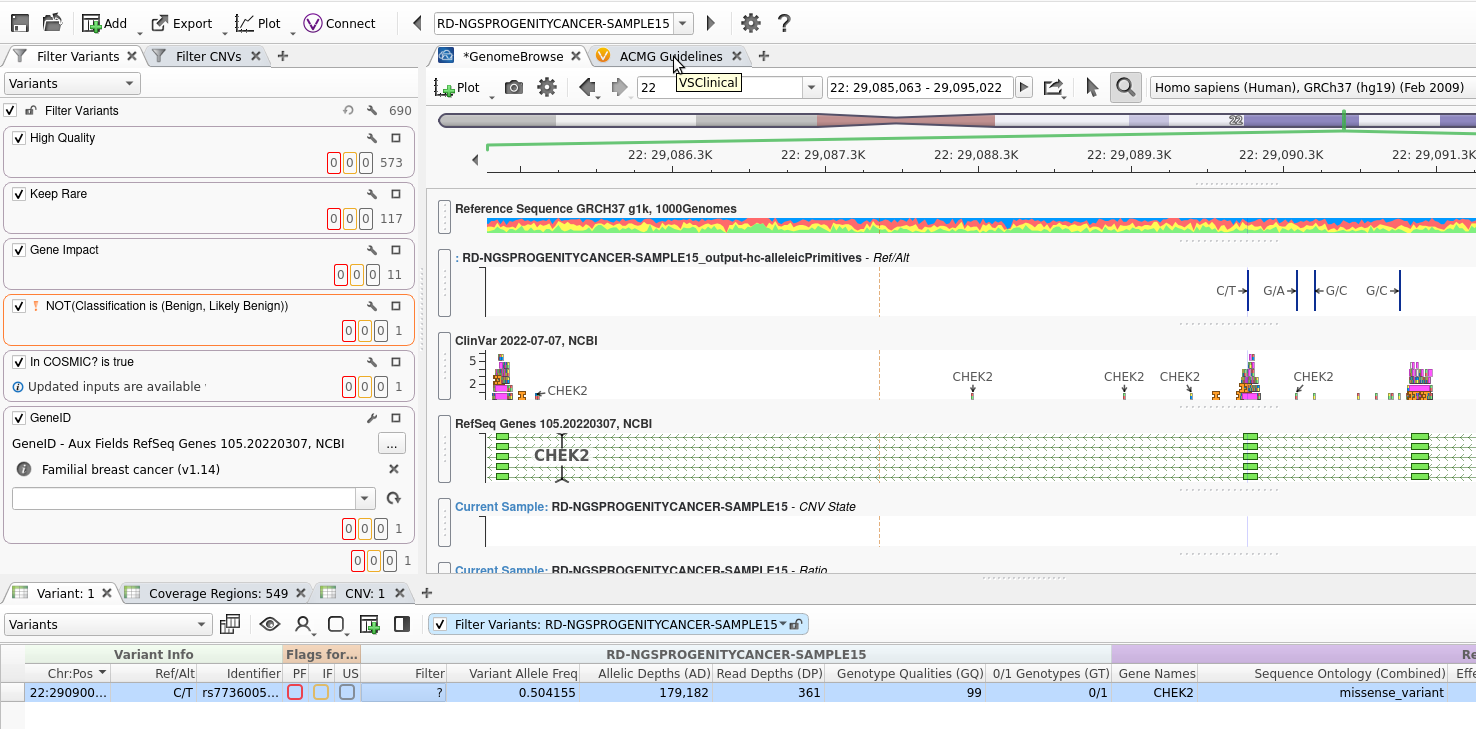
I hope you enjoyed this step-by-step review of how easily you can automate the creation of complex projects. Our example project not only housed a complicated filter chain but performed CNV analysis and annotation. Additionally, we had a Sample_Manifest that brought in sample-specific information for the VCF. All this was done with a few commands and can scale from the one example project to many more.
By automating through Sentieon and VSPipeline, you can radically increase your productivity with only minimal increases in your active time. But of course, this is just one example. For more information on increasing efficiency and lab profitability, please check out our recent webcast Maximizing Profitability in Your NGS Testing Lab presented by Golden Helix’s Andreas Scherer, CEO and President, and Gabe Rudy, VP of Product and Engineering.
Why Use VSPipeline for Your Clinical Reporting?
We’ve been trusted by doctors and scientists around the world to deliver reliable, accurate interpretations at scale. Our software is built from the ground up to be compatible with any existing lab and to deliver results with convenience, accuracy, and ease of use in mind. Whether you’re an existing VarSeq customer who’s still learning about our VSPipeline add-on or you’ve found us on your search for a new workflow automation tool, take a look for yourself. Book a demo today!
Increase your productivity and efficiency with VSPipeline!



