VSClinical users can interpret and report genomic mutations in cancer following the AMP guidelines which we’re demonstrating in this “Following the AMP Guidelines with VSClinical” blog series. Part I introduced the hands-on analysis steps involved in creating a high-quality clinical report for targeted Next-Generation Sequencing (NGS) assays. We reviewed sample and variant quality, including the depth of coverage over the target regions by the sequencing performed for each sample. Now, we are ready to move on to the next steps of the cancer workflow for creating our high-quality clinical report.
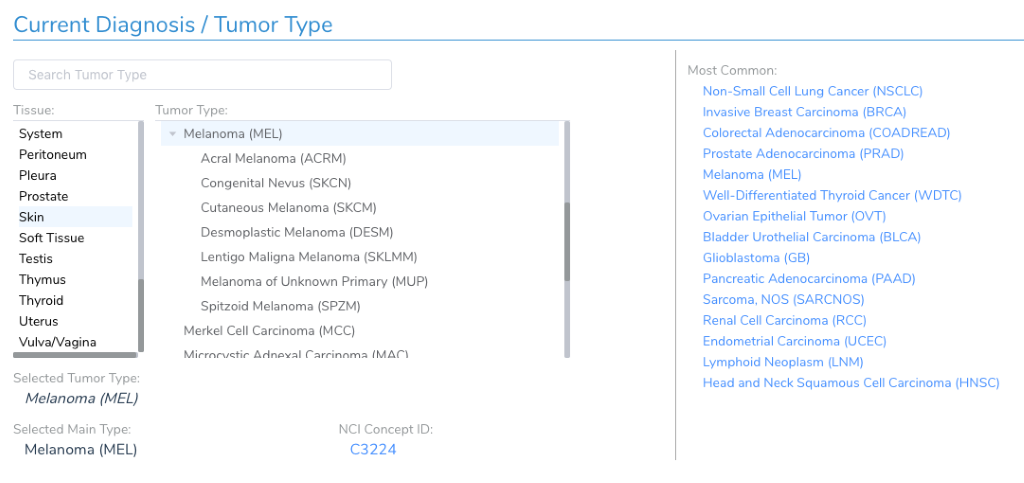
Current Sample Diagnosis
While it may seem like just another optional attribute about a patient and sample, the current diagnosis and tumor type is a critical piece of data used to follow the AMP guidelines. When it comes to evaluating clinical evidence, the AMP guidelines discuss the evidence tiers in the context of the current sample’s tumor type. For example, there may be FDA approved drugs for BRAF V600E in Melanoma, but that biomarker would not be Tier I if the current patient is diagnosed with lung cancer – rather, it would be Tier II.
Therefore, you will want to set the current diagnosis before adding any variants in the workflow so that variants are evaluated in the appropriate context.
The Full Picture: Mutation Profile
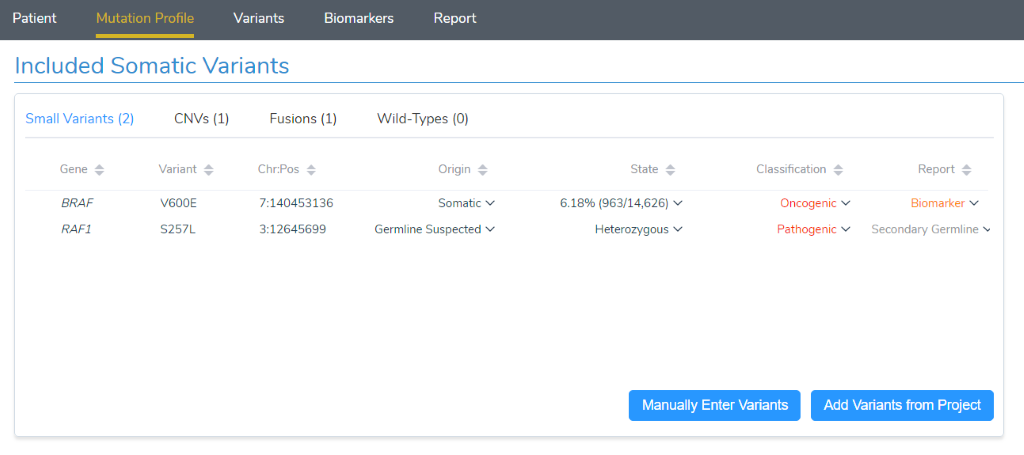
With the sample’s quality reviewed and tumor type set, we can move on to the “Mutation Profile” tab. Here we get a complete picture of what is being assessed for the current sample. This includes small variants such as SNPs and InDels, as well as CNVs, gene fusions and even Wild-Type (non-mutated) genes relevant to the current tumor type. Variants of each of these types have the potential to be evaluated as clinically relevant and make it to the final report. Let’s review each of them and their potential impact on clinical care:
- Small variants: Small variants can activate oncogenes or deactivate a tumor suppressor gene, making them relevant biomarkers for targeted therapies as well as prognostic and diagnostic markers.
- CNVs: CNV analysis can reveal whether many copies of an oncogene indicate activation, while tumor suppressor genes should be checked for single-exon to full gene deletions.
- Fusions: Oncogenes may be activated by having a fusion partner with another gene. The primary gene to interpret for clinical evidence is the oncogene, and some targeted molecular therapies list gene fusions such as BCR-ABL1as indications for use.
- Wild-Types: Certain cancers are so commonly driven by mutations in a specific gene, that having a Wild-Type of that gene provides actionable clinical context. Similarly, some targeted therapies are only applicable to tumors with specific genes as WT.
We should also review how to add a variant from our project into the mutation profile:
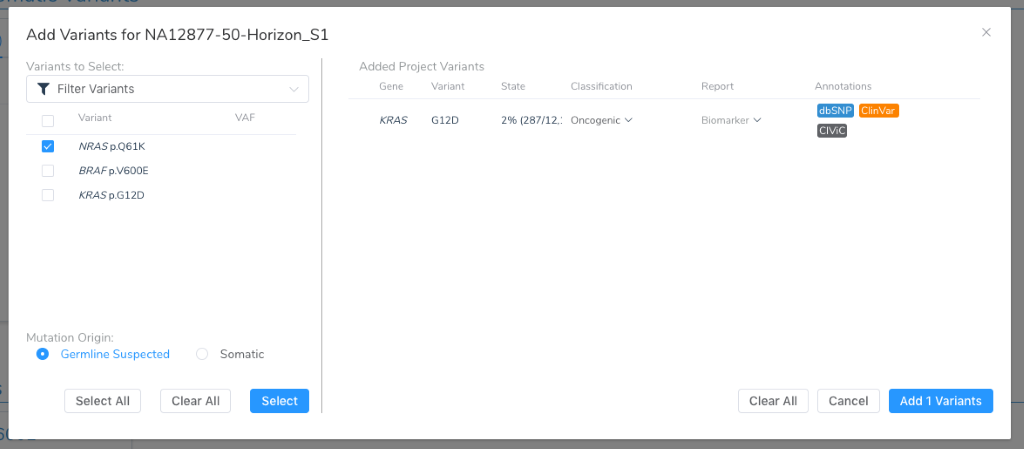
Note that at this point, the mutation origin must be specified, declaring this variant as being suspected as germline or a somatic mutation unique to the tumor. VarSeq supports tumor-normal analysis paired as well as tumor-only annotation and filtering workflows that will be able to germline from somatic variants. Regardless of how it is determined, this branch in the road will determine the subsequent analytical goals:
- Somatic: Evaluate the impact of the variant on the gene in the context of cancer, choose to move it either it forward as a biomarker or classify it as a VUS.
- Germline: Follow the ACMG guidelines for interpreting this variant as a potential Pathogenic variant for a Mendelian disorder. In most cases, report the variant in the “Secondary Germline” section of the clinical report. Certain germline variants may be an indication for specific targeted therapy and should instead be reported and interpreted as a cancer biomarker.
Automating Variant Classification
The primary hands-on interpretation of VSClinical’s AMP guideline workflow is about evaluating the clinical evidence for variants, CNVs, fusions, and Wild-Types as potential biomarkers. Many variants added to the Mutation Profile will be well established as being a driver mutation and relevant Biomarker. Others may have strong evidence of being Benign. To help make this discernment, we automate the application of our Oncogenicity Scoring Algorithm to every added variant, providing an immediate variant classification ranging from Benign to Oncogenic. In the above figure, we can see that KRAS G12D has been classified as Oncogenic.
The “Variants” tab can be used to understand the supporting evidence and scored criteria behind these auto-classifications. For variants with less presence in existing somatic catalogs or a classification of unknown significance, this tab provides supports a deep dive into all the annotations and variant-type specific evidence to make your own assessment. For germline variants, the ACMG guidelines can be followed to evaluate the pathogenicity of a variant. For variants with a somatic origin, this tab provides a similar criteria-scoring system designed to classify variants that activate oncogenes or disrupt tumor suppressor genes — a key step in any somatic analysis workflow.

Oncogenicity Scoring
The Golden Helix Oncogenicity Score was developed to provide a criteria-based scoring system similar to the ACMG guidelines but with the numeric pathogenicity scale introduced by Invitae’s Sherloc scoring system. Many of the scoring criteria are similar if not identical to those used by the ACMG guidelines, while others are specialized to match somatic annotations and clinical evidence. For example, missense variants are checked against the cancer hotspot annotation as well as active binding sites as these are often present for activating missense mutations. The criteria and strengths were developed in consultation with the GA4GH Variant Interpretation in Cancer Consortium (VICC).

Note that the scoring metrics that push a variant to the benign side of the scale are identical to the ACMG guidelines, meaning a Benign or Likely Benign classification is agnostic of whether the variant is somatic or germline.
To learn more about this scoring system, I recommend watching our Oncogenicity Scoring in VSClinical webcast recording where we cover all of this, and more!
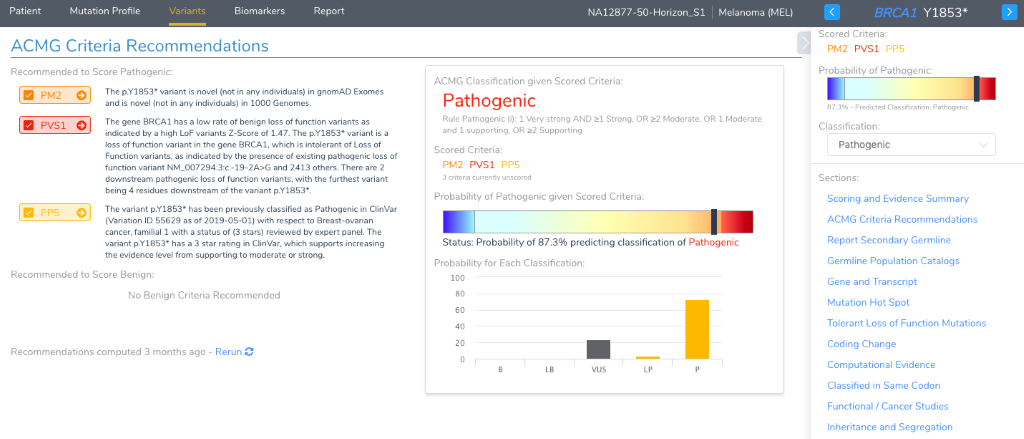
ACMG Scoring
Of labs surveyed as part of the AMP Guidelines Paper, over two-thirds report potential germline variants as part of the cancer genetic test. The paper also recommends following the ACMG guidelines for the interpretation of germline variants. Given that VSClinical was first released with stand-alone ACMG guideline support, the cancer workflow embeds this capability when the origin of a variant is selected as germline. There is a considerable amount of this workflow automated by the recommendation engine of VSClinical, with the recommended criteria brought to your attention at the very top of the “Variants” details section for a germline variant. Each criteria also has its own section, complete with supporting annotations presented in interactive visualizations. And just like VSClinical, a catalog of germline variants is saved by your lab and interpretations re-used in future samples.
Conclusion
Now that we have shown how the sample mutation profile can be built out and variants evaluated as somatic or germline, we can get into the meat of the cancer test with the interpretation of the clinical evidence for the biomarkers present in the mutation profile. Check back for Part III of this series to continue reading. We hope you’ll join us for our upcoming webcast, “Efficiently Following the AMP Guidelines with VSClinical and Golden Helix CancerKB” next Wednesday, September 11th at 12:00 PM EDT!



