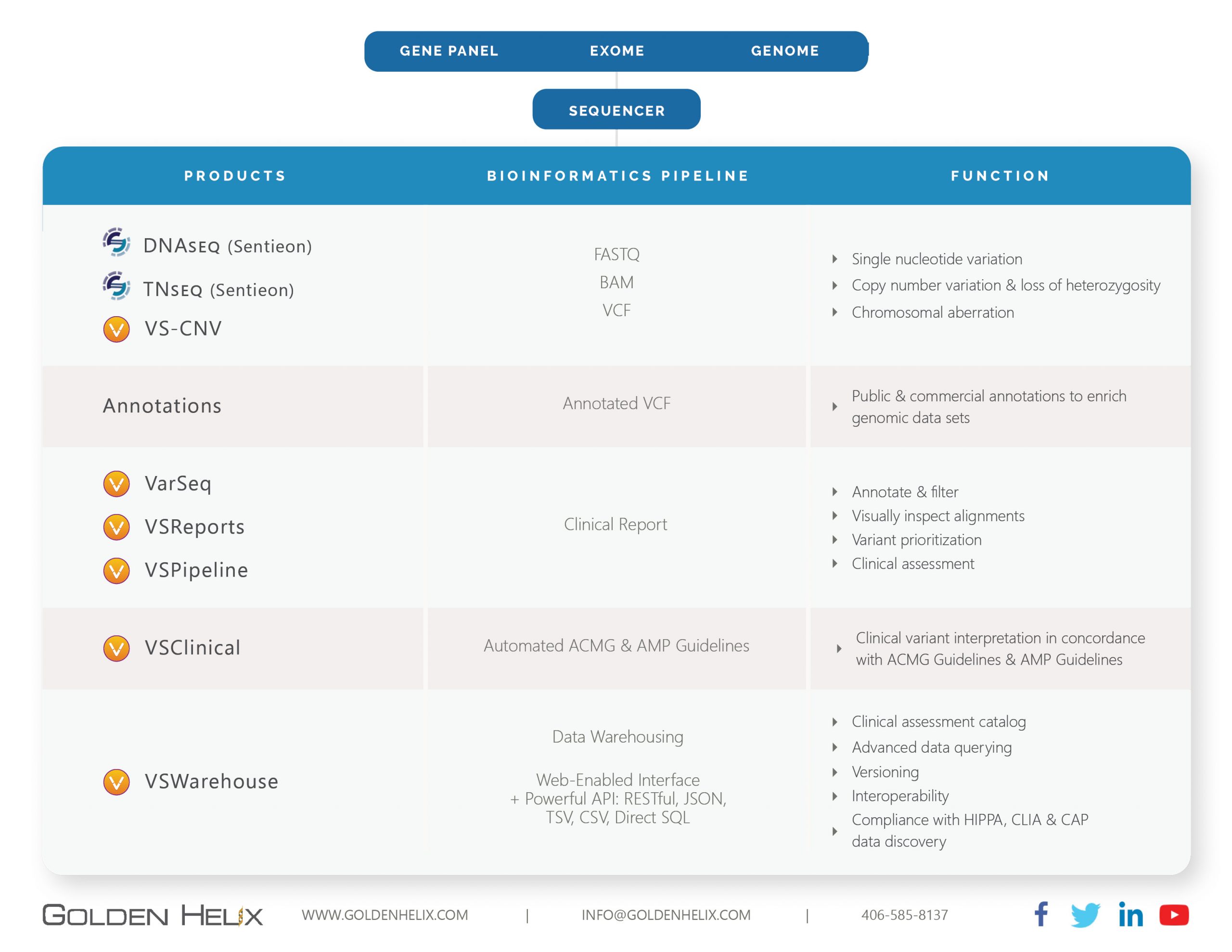
Next-gen sequencing (NGS) comprises many sophisticated steps that are often compressed into three major sections: library prep, sequencing, and data analysis. Understanding each stage of NGS data analysis is essential for building efficient clinical workflows. Obviously, the goal is to simplify each of these steps, but more often than not, there is a need for multiple tools to complete each one. Regarding the data analysis, Golden Helix seeks to provide simple yet comprehensive solutions that handle every stage from FASTQ file to clinical report with the VarSeq Suite. This blog aims to expose our readers to the different tools we provide or have developed to diminish processing time in the data analysis stage. This will begin with our partnership with Sentieon to provide a secondary solution, then discuss all the tools Golden Helix has developed to streamline the tertiary analysis. A great rundown of the tertiary solutions Golden Helix provides can be seen here.
Sentieon: Secondary Solution (FASTQ to BAM/VCF)
Sentieon’s solution seeks to be an improved drop-in replacement for BWA-MEM, GATK, Mutect2, and large cohort calling. The major improvements are maintaining the well-established accuracy in these tools and improving turnaround time for processing while maintaining 100% consistency in results. Preferentially, you will want to have a Linux environment to install this package and have adequate processing power plus memory to run samples quickly. Sentieon’s process begins with the FASTQ file, which will be aligned to the preferred genome assembly and result in the aligned reads output in the BAM file. Next, the variant caller will count alleles at various positions spread across the stacked reads in the BAM and produce the list of called variants in the VCF file. Once the output BAM and VCF are generated, the files are imported into our tertiary solution VarSeq.
VSPipeline
Golden Helix has developed a command-line interface for running batches of samples for automated project creation for the advanced application of the tertiary stage. The essence of VSPipeline is to speedily create completed projects in VarSeq (discussed in detail below), where the user starts their analysis on the filtered clinically relevant variant. VSPipeline is largely dependent on constructing a project template in VarSeq that is designed to be routinely applied to batches of samples over time. Rapid project completion is achievable in a decent computational environment that can start with the FASTQ and end at a completed project prepped for report rendering with CNV computation in approximately 30 minutes for a single exome sample (based on Golden Helix internal run times).
VarSeq
VarSeq is the graphic user interface tool that handles the import of full VCF content and necessary sample data relevant for clinical reporting. There are many tools in VarSeq, including GenomeBrowse for visualization, VSClinical for variant evaluation following ACMG/AMP guidelines, and reports. Fundamentally, VarSeq is also handling the entire variant filtration step to isolate the clinically relevant variant. VarSeq scales for all gene panel, whole exome, and whole genome level data. The initial goal when getting started with VarSeq is to develop a workflow template that will be validated and deployed across multiple sample batches. The template filter is composed of many annotations and algorithms hosted in the VarSeq software. Common filtering strategies may incorporate:
- Variant quality fields like read depth or genotype quality
- Removing common variants with population catalogs like 1KGPhase3 or gnomAD exomes/genomes
- Prioritization of variant impact on gene with RefSeq
- Panel or Phenotype based searching
- ACMG classification
VarSeq is also the environment where users can perform CNV analysis for panels, exome, and whole genome data by leveraging the coverage data stored in the sample BAM files. Whether for CNVs or standard SNV/indel output in the VCF, once the user has filtered to the variant of interest, the next phase is to handle the variant evaluation in VSClinical.
Give VarSeq a try with the VarSeq Viewer!
Visit our site here for more info
Explore pre-built projects to interact with the annotations and filters, visualize selected variants with pile-ups, and export data to widely used formats.
VSClinical
VSClinical serves as the variant interpretation hub to handle the comprehensive review of all variant evidence and capture appropriate ACMG and AMP guidelines. In this process, VSClinical supplies the user with automated recommendations. In other words, our software will display all variant criteria with automated prompts to simplify the evaluation. However, the user still has the final say on classification plus consideration for any comments/caveats. This includes our recent upgrade to process CNVs via ACMG guidelines considering known gene dosage sensitivity in ClinGen. Once the variant is evaluated, interpretation drafted, and classification reached, all variants are stored in assessment catalogs to ease re-evaluation on future samples sharing the variant and potential changing classifications. Additionally, VSClinical is the environment where the user will carry all primary and secondary findings into a custom rendered clinical report. At the end of the day, all completed projects, variant knowledge, and reports need to be stored in an environment where the data can be easily queried and leveraged for future sample projects.
VSWarehouse
VSWarehouse is the solution for storing all your genomic data processed in VarSeq. Leveraging this data can be beneficial in several ways. The entire cohort of sample variants are uploaded into a server, and the corresponding variant allele frequencies prove useful in VarSeq workflows. One such filter is to remove common variants seen among the cohort that are likely artifact variant leftover from the secondary analysis stage. Additionally, users may wish to extract all detected variants across a single or list of genes or perhaps known pathogenic variants seen in databases like ClinVar. VSWarehouse will store your project data with allele frequencies and store the assessment catalogs for ACMG/AMP-based interpretations and classifications and the potential to store your custom clinical reports. Not only is this content stored, but it is also accessible for all licensed users to share the genomic content and unify the genomic knowledge base while dramatically improving workflow efficiency.
Obviously, this is meant to be a high-level overview of each tool. The main goal was to give our readers context on the available solutions on our software menu. Essentially, once the sample data comes out of the sequencer, we have the complete list of all necessary tools to complete the analysis and reporting process. Some or maybe all of these tools would be relevant to you. In that case, we’d be more than happy to set up a demonstration of the desired tools.



