Before examining the clinical evidence associated with a specific mutation, a clinician must establish that the variant is likely to be a driver mutation which generates functional changes that enhance tumor cell proliferation — a foundational step in variant interpretation. Our recent blog series “Following the AMP Guidelines with VSClinical” briefly mentioned how the oncogenicity scoring system in VSClinical could be used to automate and assist the choice of which variants are worth reporting and evaluating as a biomarker.
In this blog, we will dive deeper into this topic and explain VSClinical’s capabilities for determining the oncogenicity of a variant. I will also cover our oncogenicity scoring system and the various criteria used in variant classification to distinguish driver mutations from benign variations and variants of uncertain significance. If this is a topic of interest for you, I recommend watching my on-demand webcast recording on this “Oncogenicity Scoring in VSClinical“:
After using VarSeq to filter down to a small set of interesting variants, VSClinical’s oncogenicity scoring system can be used to examine whether a variant is likely to be a driver mutation. Our oncogenicity score is an additive scoring system, with scores exceeding ‘3’ indicating an ‘oncogenic’ or ‘likely oncogenic’ effect. When you first open the oncogenicity scoring pane, you will be greeted with a basic overview of the variant, including the variant’s genomic position, gene, and a summary of the relevant evidence.
The next section includes a list of the individual recommended scoring criteria along with an oncogenicity classification. The variant shown below is NRAS G12D, a textbook example of a driver mutation. The first thing to notice is that this variant has been classified as ‘oncogenic’ with a score of ’10’, which is well-above our threshold for classifying a variant as oncogenic.

In the left panel, we can see the specific pieces of evidence that were used to obtain this classification. For example:
- We can see that the variant occurs in nearly a thousand samples in COSMIC,
- It has been previously classified as pathogenic in ClinVar,
- It is predicted to be damaging by our in-silico prediction algorithms,
- It occurs in cancer hot spots as well as an active binding site,
- Seven other variants within six amino acid positions of our variant of interest have been shown to be pathogenic.
For any of these criteria, we can easily examine the data used to determine its applicability. For example, if we click on the arrow to the right of the in-silico prediction recommendation, we will be taken to a detailed description of the criteria and the evidence used to support it. Here we can see the scores for both SIFT and PolyPhen predicting that the variant is damaging. We are also presented with a multi-species alignment for the relevant region, allowing us to confirm that the variant is conserved across all mammalian species.

The next criteria I would like to examine is NP+1. This criterion is applied to missense variants for which there are multiple nearby pathogenic missense variants, but none that are classified as benign. We are currently being recommended this criterion because seven variants within six amino acid positions of our variant of interest have been shown to be pathogenic, while none have been shown to be benign. We can verify this recommendation by examining the table of nearby variants.
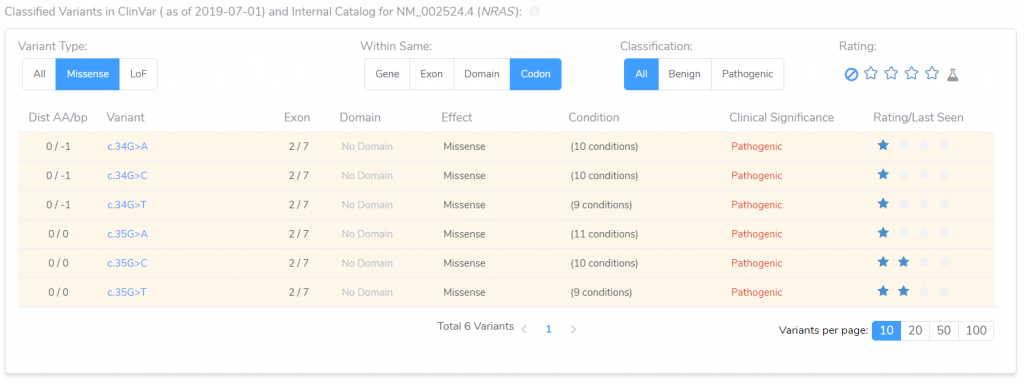
This table presents all variants in both ClinVar and the clinical lab’s internal catalog. It includes information about each variant’s effect, and its position relative to the variant of interest. By applying a couple of filters, we can quickly confirm that there are six variants in the same codon as our variant of interest, all of which have been classified as pathogenic, thereby establishing the applicability of the NP+1 recommendation.

Now that we have examined a straightforward oncogenic mutation, we will take a look at a variant that is a bit more complex. Specifically, we will examine a mutation in MLH1 that is 5 base pairs into the intron of the gene. Generally, such an intronic variant would be ruled out as benign, but this variant has instead been given a classification of likely oncogenic.

To understand what is going on, we will need to examine the two pieces of evidence used to reach this classification. First, the variant is in a highly conserved region. In fact, if we look at our multiple species alignment, we can see that this particular locus is conserved all the way down to Lamprey. While this is unusual for a variant in an intronic region, if we examine out splice site predictions, we can clearly see what’s going on, as all four of our splice site prediction algorithms are predicting this variant will disrupt an existing splice site.
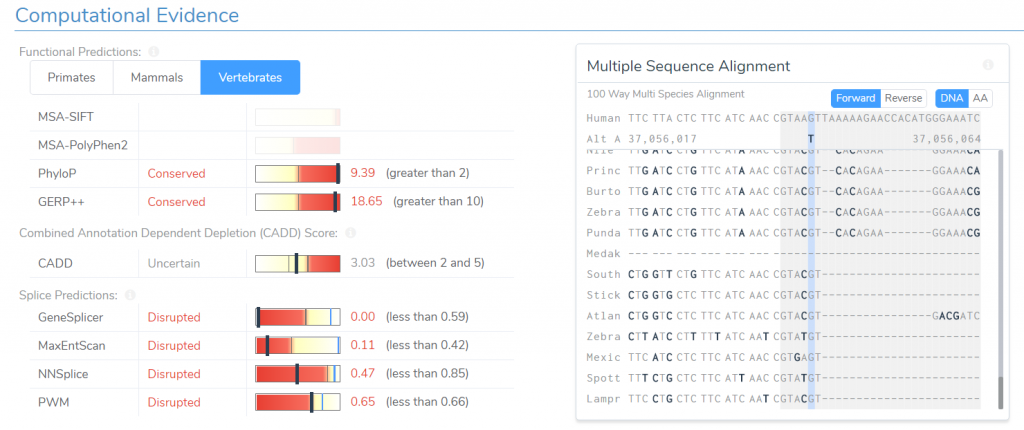
Normally, we would consider a variant’s presence in an intronic region to be evidence of a benign effect, but because all of our splice site prediction algorithms predict this variant to disrupt an existing splice site, we no longer consider this evidence for benign impact. Instead, the variant’s effect on the existing splice site is considered moderate evidence for oncogenicity.
It is worth noting, however, that the presence of only one of these pieces of evidence, would not be sufficient to recommend a ‘likely oncogenic’ classification. That is if the region was highly conserved, but we had no evidence that the variant impacted splicing, then this would result in a variant of uncertain significance. Likewise, if the variant was predicted to impact splicing, but the region was not conserved, then this variant would again be classified as having uncertain significance. It is only with both of these pieces of evidence together that we can determine that this variant is likely to be oncogenic despite the fact that it occurs in the intronic region of the gene.
The final variant that we will examine today is a frameshift mutation in the gene MUC4. Notice that this variant has been classified as likely benign, despite the fact that this is clearly a Loss of Function (LoF) mutation. At first glance, this appears to be an unexpected result.
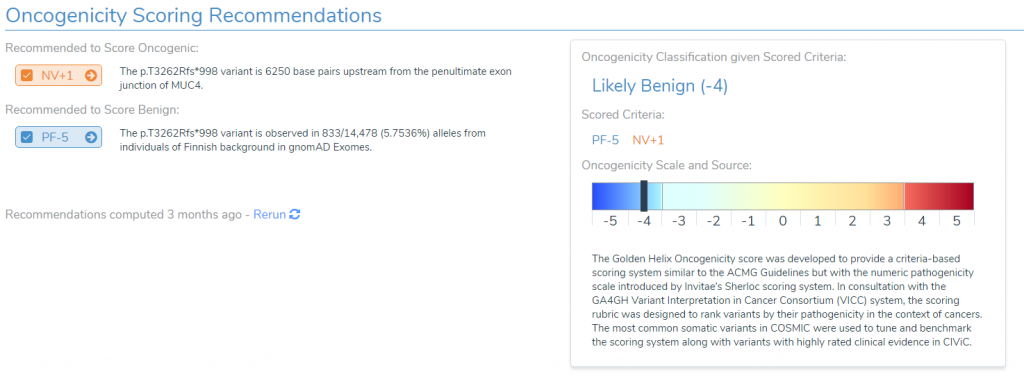
This variant has two conflicting pieces of relevant evidence. The first is evidence for oncogenicity, due to the fact that this is a loss of function variant which is over 6,000 base pairs upstream from the penultimate exon. Normally we would expect this to be a damaging mutation, but the gene MUC4 is actually an oncogene, thus in order to be classified as oncogenic, we would need evidence that the variant is an activating mutation for the gene. However, loss-of-function variants make up less than 1% of all variants in this gene in COSMIC, providing evidence that loss-of-function variants are not a mechanism for activating this gene.
The above evidence is enough to prevent us from classifying the variant as oncogenic, but in order to reach our classification of likely benign, we need one last piece of evidence, namely PF-5. This criterion is applied for variants that are common in at least one population catalog and is considered strong evidence for a benign classification.
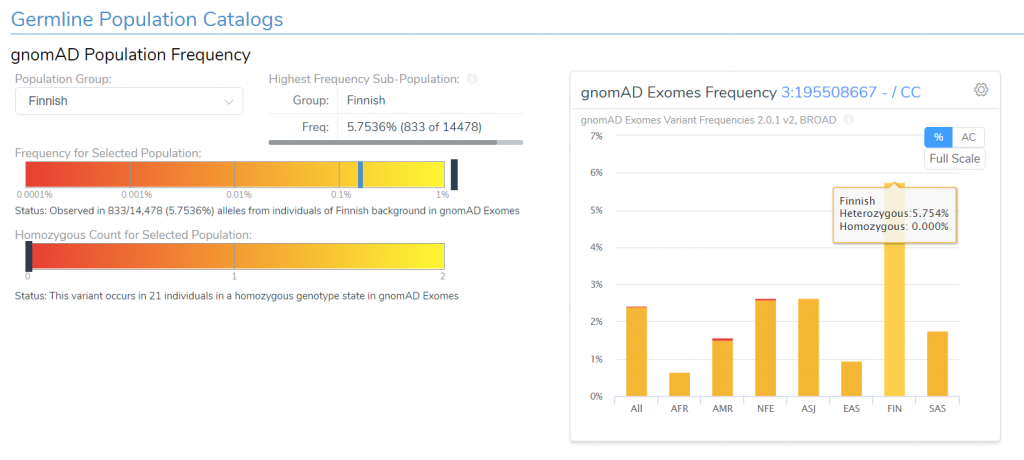
Looking at the plot of our population frequency data from GnomAD, we can see that this variant occurs at around 2.4% in all individuals in a heterozygous state, and it occurs at around 6% in the Finnish sub-population. This frequency is much higher than we would expect for a truly oncogenic variant, thus, despite the variant’s loss-of-function effect, we have good reason to believe that this variant is likely benign and can, therefore, be excluded from interpretation.
Hopefully, this blog post has shed some light on our oncogenicity scoring system and demonstrated how VSClinical can be used to distinguish between benign variants and oncogenic mutations that drive tumor cell proliferation. VSClinical is part of Golden Helix’s broader suite of oncology software designed to support somatic interpretation workflows from panel to whole-genome scale. If you have any questions or comments about the information presented here or about our software, please don’t hesitate to reach out and contact us.



