You probably haven’t spent much time thinking about how we represent genes in a genomic reference sequence context. And by genes, I really mean transcripts since genes are just a collection of transcripts that produce the same product.
But in fact, there is more complexity here than you ever really wanted to know about. Andrew Jesaitis covered some of this in detail as he dove deep in the analysis of variant annotation against transcripts in his recent post The State of Variant Annotation: A Comparison of AnnoVar, snpEff and VEP.
The databases that catalog and provide labels for transcripts are really cataloging transcribed RNA, like you might identify from a RNA-Seq experiment, as well as the spliced and translated protein products. To represent these RNA sequences in genomic space we must actually take a step backward. In fact, it takes a special type of alignment algorithm (see my webcast on alignment algorithms, the same family of algorithms are applied to this problem).
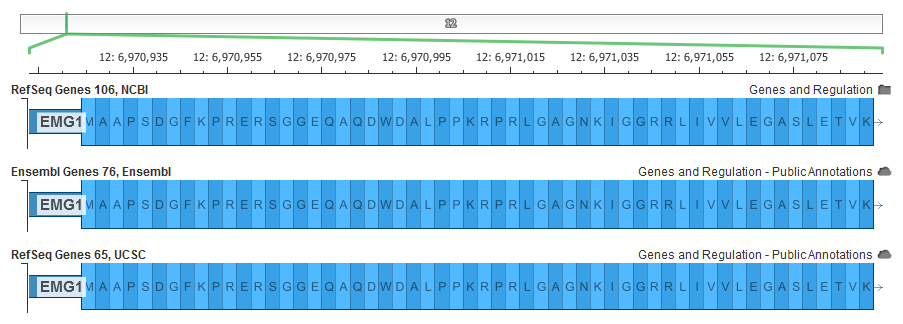
Well it turns out these algorithms do not always agree on the “best” alignment. Here is a very poignant example of their disagreement:

First we have our new RefSeq Genes track that I will be discussing in this post. This is built off of a GFF file that is part of the NCBI Human Annotation Release 105 using NCBI’s Splign algorithm. The second track is the latest Ensembl release on GRCh37, which uses the Ensembl genebuild algorithm, and finally the UCSC alignments of the same RNA using their BLAT algorithm.
You can see they disagree on how to handle a “gap” between the NM_006331 transcript’s RNA sequence and the human genomic sequence. The Ensembl alignments most correctly preserve the actual protein sequence of NP_006322.4 in this case by introducing a 2bp intron (which has no real biological reality). Note that given this is in the first exon, this disagreement extends for the rest of the protein sequence.
Of course, they are trying to do the best with a bad situation where the reference sequence is not ideal. In fact, this is a great example of the real improvements of the latest GRCh38 reference sequence. It incorporated a “patch” that changes the local sequence at this region. You can see the same gene with all three algorithms agreeing on simple alignment with the GRCh38 genomic reference:
These types of issues are certainly not prolific, but do enough analysis and you will run into them and discover how they can result in dramatic differences in how a variant is annotated.
In this example, the Splign and BLAT algorithms place exon 12 in NM_052814.3 at completely different locations:

A variant falling in one of those locations would have its classification differ between coding and intronic dependent on which annotation source you were to use.
No Best Choice
Is one algorithm empirically better than the rest? It turns out you can find examples of each algorithm doing a subjectively interpretable “better” job under different circumstances.
We spent some time talking to Reece Hart of Invitae recently, and he referred us to his slides on this exact topic which I recommend you review if this interests you.
Changing our Default Gene Track
In our latest release of Golden Helix SVS 8.2.0 and GenomeBrowse 2.0.4, we shipped by default the RefSeq Genes from NCBI Human Annotation 105 as the default gene track. Going forward we will curate RefSeq genes from NCBI directly and no longer from UCSC.
There are a few of reasons we believe this to be the right path going forward:
- NCBI provides mitochondrial gene mappings to NC_012920.1; the MT referenced used in GRCh37_g1k and GRCh38 that nearly all NGS alignment is done with.
- NCBI has official releases of annotations for species, compared to UCSC more continuously changing approach.
- NCBI provides extremely good cross-references for their transcripts with other resources, providing great hyperlinks and summary information for analysis.
- The NCBI set of alignments includes more non-coding transcripts such as microRNA, tRNA as well as various annotations of transcripts such as “pseudo” and “predicted”.
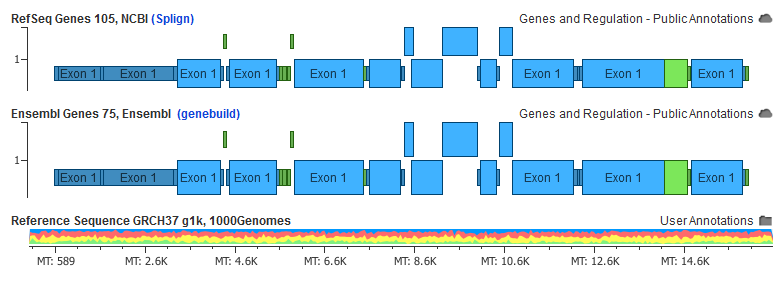
Mitochondrial Genes
The UCSC provided RefSeq genes does not actually provide any mitochondrial gene annotations. As inherited mitochondrial gene disorders are testable with NGS gene panels, it’s important to provide as much annotation support for Dx tests based on variant data.
The Revised Cambridge Reference Sequence (rCRS) has been adopted by the community (and now by GRCh in 38) as the standard reference to use, and probably would have been used by UCSC if it was available when they defined their “hg19” reference (and decided not to update hg19 when it was clear that rCRS was the way forward).
You can see that the RefSeq transcripts for “MT” in the new NCBI track are identical to those provided by Ensembl as these are now very stable annotations:
To support this switch, we also updated our default genome assembly for new projects to GRCh37_g1k (previously it was based on the UCSC hg19) in the latest SVS and GenomeBrowse releases.
Full Transcript Set
We plan to consistently support the approach of having a full set of RNA products represented in our gene track with predicted mRNA, pseudo (incomplete) RNAs and non-coding RNAs all intermixed. To achieve this, we took great care to provide extra fields to the track that allow for easy identification and filtering of the type of transcript you are interested in.
In this way, we updated our Variant Classification algorithm in SVS 8.2.0 to by default only annotate non-predicted, protein-coding transcripts (that will have NM_ transcript identifiers). This brings RefSeq in line with the Ensembl gene track which similarly has been inclusive of incomplete and predicted transcripts.
Here are the stats of the RefSeq Genes 105 transcript set by type:
| Transcript Type | Count |
| mRNA (NM_*) | 34,663 |
| mRNA Predicted (XM_*) | 30,077 |
| microRNA | 1,501 |
| ncRNA (rRNA, tRNA, other) | 7,090 |
| Puedo, incomplete | 11,619 |
| Total | 84,950 |
Cross Reference Database Fields
While I could write many posts on the difficulties of dealing with the GFF file NCBI produced, it was worth the effort to handle the many edge cases which demanded special parsing to produce our latest default gene track. The extra annotations provided by NCBI have been cleaned up and hyperlinked to provide a platform for exploring a gene and its relevance to your data.
Below is our Console output for a transcript with a full set of links to various databases related to it:
| Gene Name | PMPCA |
| Transcript Name | NM_015160.1 |
| Type | mRNA |
| Transcript Type | mRNA |
| Product ID | NP_055975.1 |
| Protein ID | NP_055975.1 |
| GeneID | 23203 |
| HGNC | 18667 |
| MIM | 613036 |
| CCDS | CCDS35180.1 |
| HPRD | 07500 |
| Summary of Product | mitochondrial-processing peptidase subunit alpha precursor |
Taking Data Curation Seriously
We spent much of the last month making many iterations over this gene track, and it’s just one of the many data sources we are committed to providing in our free annotation library to our SVS and GenomeBrowse users.
Now that we codified our logic into a script, you can actually curate your own NCBI gene track for a different species or build you may be interested in from the SVS Data Source Library under the Utilities menu.

Interesting post.
Any recommendations on some good tools that convert gff to gtf? A lot of the tools and algorithms out there accept only gtf files as input. I have been struggling with the gff files. I am using a genome that does not have a ready made gtf file.
Because GFF is very general in terms of what you can put in it’s hierarchical structure, there can really be no single tool to convert GFF to GTF. It is not that surprising then that most tool authors prefer to support GTF, as it is ]relatively constrained in terms of how a transcript alignment is described.
We spent nearly a whole month building a custom script to pull out relevant, but not uniformly encoded data out of the NCBI Annotation GFF files. If you want to convert that specific format of GFF, you could use our tool in SVS and save back out to TSV (which will be pretty close to GTF). We may add support to saving to GFF as well in the future.
Hello!
Very cool post. It’s interesting how annotations for the same gene and similar transcripts differ across the different annotation groups. I just wanted to post here that the UCSC Genome Browser (whom I work for) just released a set of annotation tracks based entirely on coordinates and alignments provided by NCBI’s RefSeq group. We also still have our UCSC derived BLAT-based alignments track as well, but the RefSeq -derived “Curated” subset has become our default. Here’s our announcement on it: https://genome.ucsc.edu/goldenPath/newsarch.html#030317
Hey Matt,
I think this is a great move by UCSC and makes things very consistent for interpretation.
I couldn’t find in your announcement or track documentation, but are you using the recently updated and released “interim” GFF files?
ftp://ftp.ncbi.nlm.nih.gov/genomes/Homo_sapiens/GRCh37.p13_interim_annotation/