Thank you to those who attended the recent webcast, “Exome Analysis with VS-CNV & VSClinical: Updated Strategies & Expanded Capabilities”. For those who could not attend but wish to watch, here is a link to the recording.
In this webcast, we covered the capabilities and updates that have been incorporated into VarSeq that enhance whole exome sequencing workflows. The new features and updates for VarSeq 2.2.3 that were discussed fall into three main categories:
- Updates to the CNV calling algorithm to have increased precision and enhanced quality flags ultimately reducing the number of false-positive CNV calls in exomes and large panels.
- Enhanced variant analysis to include non-coding RNA, mitochondrial, and splice site variants. Also, updates to gene annotation algorithms allow users to analyze variants that are upstream/downstream of coding regions.
- How to set up a new best-practiced workflow to incorporate the new features that have been added to VarSeq 2.2.3 like incorporating gene panels and coverage information for creating clinical reports.
Many of you asked some fantastic questions that we, unfortunately, did not have enough time to answer live. However, I would like to take the opportunity to provide some answers to those questions now!
Question: How do I change my existing project templates to detect non-coding RNAs like you demonstrated?
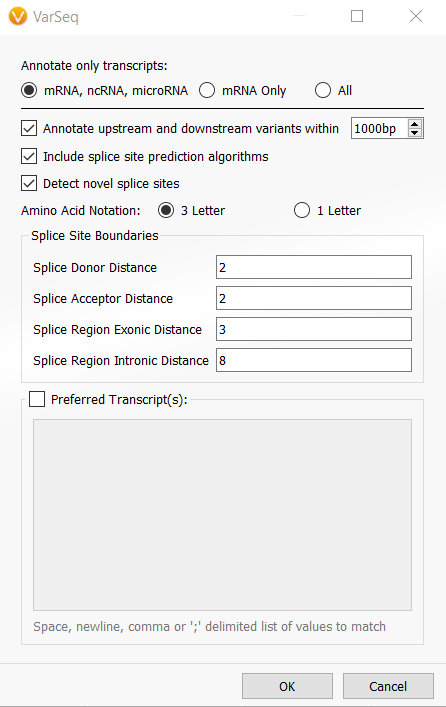
Answer: VarSeq 2.2.3 now provides users with the ability to edit the settings/options of algorithms that already exist in the project. To change the settings of the gene annotation algorithm, RefSeq Genes, just right-click on the RefSeq Genes output in the variant table and choose Edit. When editing the algorithm you can opt into detecting mRNA, non-coding, microRNA variants, and variants that are upstream or downstream of coding regions (Figure 1). Then you can save the project as a template (File > Save Project as Template…) to update the project template.
Question: Do you have any specialized annotations for Mitochondrial variants?
Answer: Yes, VarSeq currently offers MITOMAP annotations that are fully equipped with an updated list of mitochondrial variants. Typically, these variants are those that are already established as clinically relevant. However, the data source library within VarSeq provides references to the MITOMAP page wherein you can explore variants in more detail if you so choose.
Question: Have you looked at the Genome in a Bottle benchmark papers for structural variants?
Answer: Yes, we have used the Genome in a Bottle benchmarking data for structural variants as they do fantastic work benchmarking small variant calling and they just recently achieved publication status on structural variant benchmarking which is based on the HG002 sample. We used this sample for target capture exomes in addition to other microarray samples when optimizing whole exome CNV calling algorithms in VarSeq. We plan to cover the details of the benchmarking process and precision metrics in an upcoming webcast.
Question: When will this new release of VarSeq be available?
Answer: The 2.2.3. VarSeq release is in the final stages of testing so we expect to release the software within the next few weeks.
Question: Are there tools that allow us to score non-coding upstream and intronic variants that could impact regulation?
Answer: Previously, non-coding, upstream and intronic variants were not able to be evaluated with VSClinical. However, VSClinical now supports annotating and analyzing these variants with the ACMG guidelines. To hone in on the interesting variants, a good place to start is to look for variants with pathogenic submissions to ClinVar, or VarSeq has annotations available that can be used to see if the variant is overlapping regulatory elements. How these regulatory elements regulate gene expression can happen in a variety of different ways, but depending on the application, UCSC Genome Browser offers downloadable tracks that can be easily converted to annotation sources that can be used within VarSeq.
Question: Can we detect translocations and other alterations like gene inversions with this application?
Answer: Detecting translocations and similar alterations do not necessarily apply to target panels. However, there are potentially two situations wherein I can see these alterations being analyzed:
- Whole genome analysis- Looking at these translocations at a read-by-read level and looking for where the strand flips.
- Utilizing PCR kits- Essentially these kits have probes to capture the boundaries of the alteration. In cancer, this method is used for common recurrent fusion genes with known splicing locations.
The general purpose of any webcast is to showcase the capabilities of our software. If you would like to have a more in-depth introduction to all GoldenHelix solutions please contact us at [email protected]. Thank you again to all of those who attended and we look forward to having you join us for our future demonstrations.
Feel free to also check out some of our other blogs that contain important, useful news and updates for the next-gen sequencing community.



