It is common knowledge that variants can be germline or somatic depending on whether the variant was inherited or acquired after birth. A well-known example is cancer-causing mutations in the BRCA genes, wherein the mutation may or may not have been inherited. Understanding the origin of the cancer-causing mutation is important when assessing potential treatment options as well as identifying at-risk individuals. Thanks to recent advances in genomic sequencing technology, we can have a better understanding of the tumor genome and thus its impact on personalized medicine. Clinicians can now leverage information from somatic tumor testing to identify hereditary cancer risks and not have to rely on family history alone to identify at-risk individuals (1). Therefore, I want to discuss an interesting gene with mutations that are involved in germline disease and germline and somatic cancers and how VSClinical can assist in evaluating such variants according to the AMP and ACMG guidelines.
Before I jump into the software, I want to provide some background on the gene and mutations that we will be evaluating. The WT1 gene produces the Wilms tumor protein 1 which normally functions as a transcription factor and is necessary for the development of the kidneys, ovaries, and testes before birth. After birth, the WT1 protein plays a role in cell growth, differentiation, and apoptosis in the glomerulus in the kidneys (3). In terms of germline mutations that result in disease, there are at least 80 known mutations in exon 8 and 9 in the WT1 genes that have been known to cause Denys-Drash syndrome. However, more rare mutations occurring in the same exons result in Fraiser Syndrome. A larger germline deletion in the WT1 results in WAGR syndrome which results in childhood kidney cancer known as Wilms tumor, aniridia, genitourinary anomalies, and intellectual disabilities (3).
Each of these germline mutations increases the risk for Wilms’ tumor, which is the most common childhood kidney cancer (2). However, most of the mutations that cause Wilms’ tumor are actually somatic mutations. When evaluating Wilms tumor samples, it is important to note whether these mutations were germline or somatic as germline WT1 alterations have been identified in an estimated 2–4% of presumed sporadic Wilms tumor cases and would thus influence how these variants should be reported and treated (4).
Let us now take a look at a workflow that evaluates several WT1 variants in VSClinical according to the ACMG and AMP guidelines.
From a VarSeq project, I have built a standard filtering logic that will keep variants that are high quality, rare according to population frequency catalogs, pathogenic or conflicting according to ACMG classification, and are included in the COSMIC database. To specifically hone in on the variants that are in the WT1 gene, I have also included a gene list using the Match Gene List algorithm consisting of the WT1 gene exclusively, which leaves us with seven variants that we can evaluate in VSClinical (Figure 1).

Figure 1: Filtering logic that narrows our variants search to WT1 variants for evaluations according to the AMP and ACMG Guidelines.
Using VSClinical’s AMP Workflow, users can evaluate and classify variants according to the ACMG or AMP guidelines within the same interface. Before we choose the WT1 variants from our VarSeq project, we first need to specify the patient tumor type. This will ensure that all the interpretation for these variants will be in the context of Wilms’ Tumor (Figure 2).

Figure 2: Defining the patient’s tumor type within the patient profile to capture relevant variant interpretation.
I have selected 4 WT1 variants from the VarSeq project for classification and interpretation. I added these variants not only as somatic variants but also as germline suspected variants so we can compare the results from oncogenicity scoring with the AMP Guidelines and pathogenicity scoring with the ACMG Guidelines — a workflow that reflects best practices in somatic analysis (Figure 3).
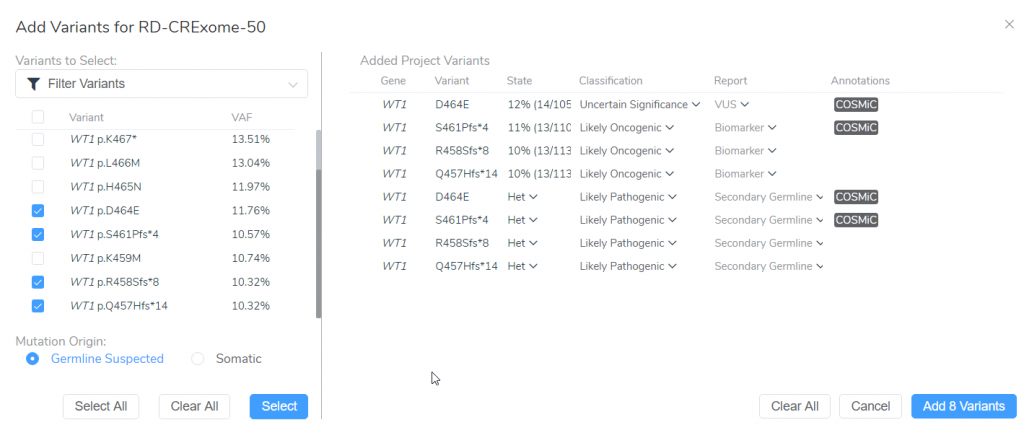
Figure 3: Adding 4 WT1 variants as germline and somatic variants from our VarSeq project for analysis with the ACMG and AMP guidelines.
To begin, I want to draw attention to the WT1 D464E variant (Figure 3). When this variant is added as a somatic variant the classification is a variant of uncertain significance. However, when the same variant is added as a germline variant, the classification is likely pathogenic. This variant highlights the importance for determining whether this patient tumor sample has a germline or somatic mutation origin as the clinical report will include differing interpretation and classification for this patient. Let’s explore these differences further.

The somatic D464E mutation is classified as a variant of uncertain significance according to 2 oncogenicity scoring criteria; this variant occurs in somatic catalogs, specifically COSMIC, and the variant is located in a mutational hot spot. This same variant is scored as likely pathogenic when evaluated according to the ACMG guidelines criteria; this variant is novel in population catalogs, it is located in a mutational hotspot, and it is a missense variant resulting in an amino acid change at the same position of a previously classified pathogenic variant (Figure 4).
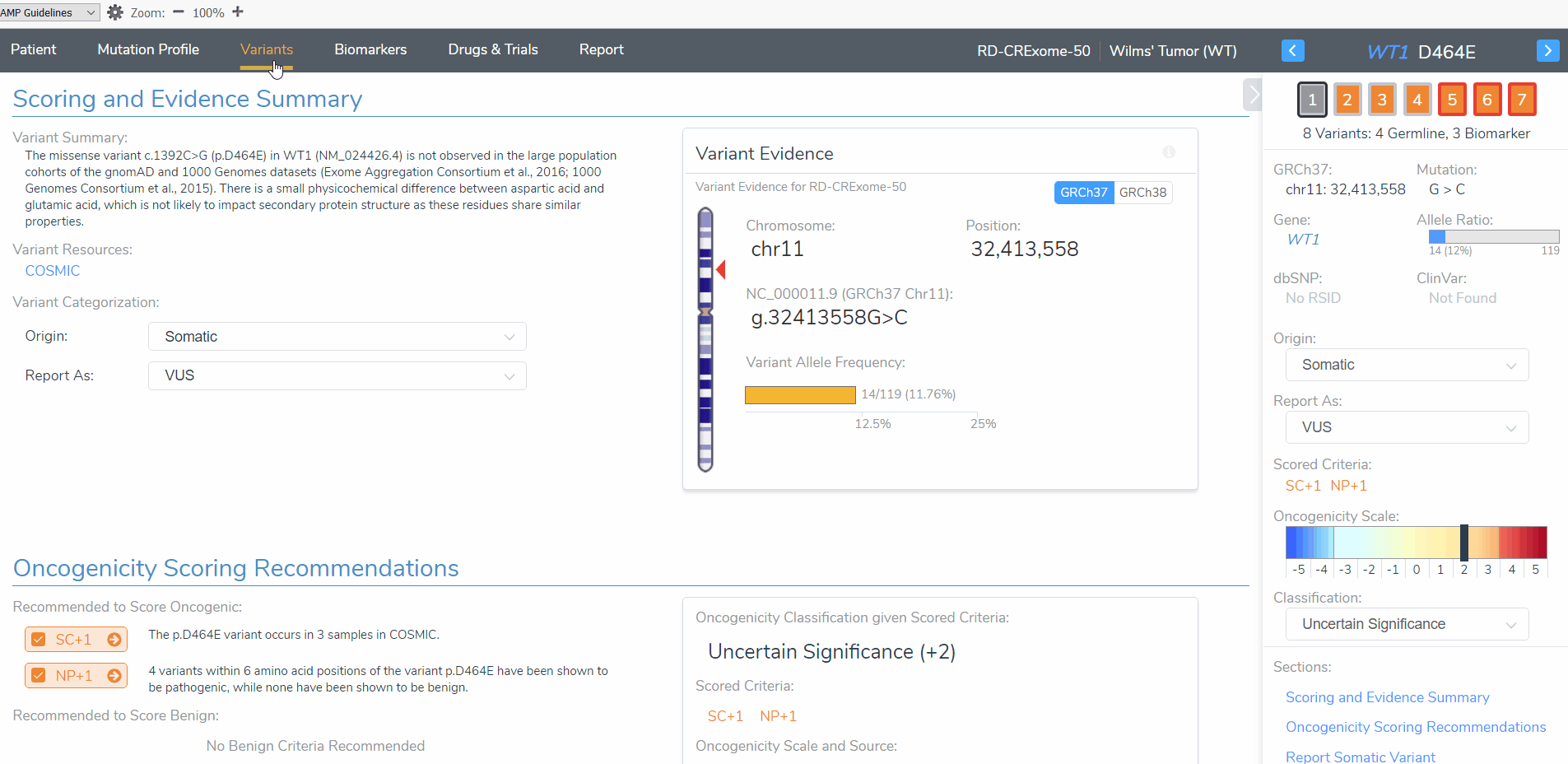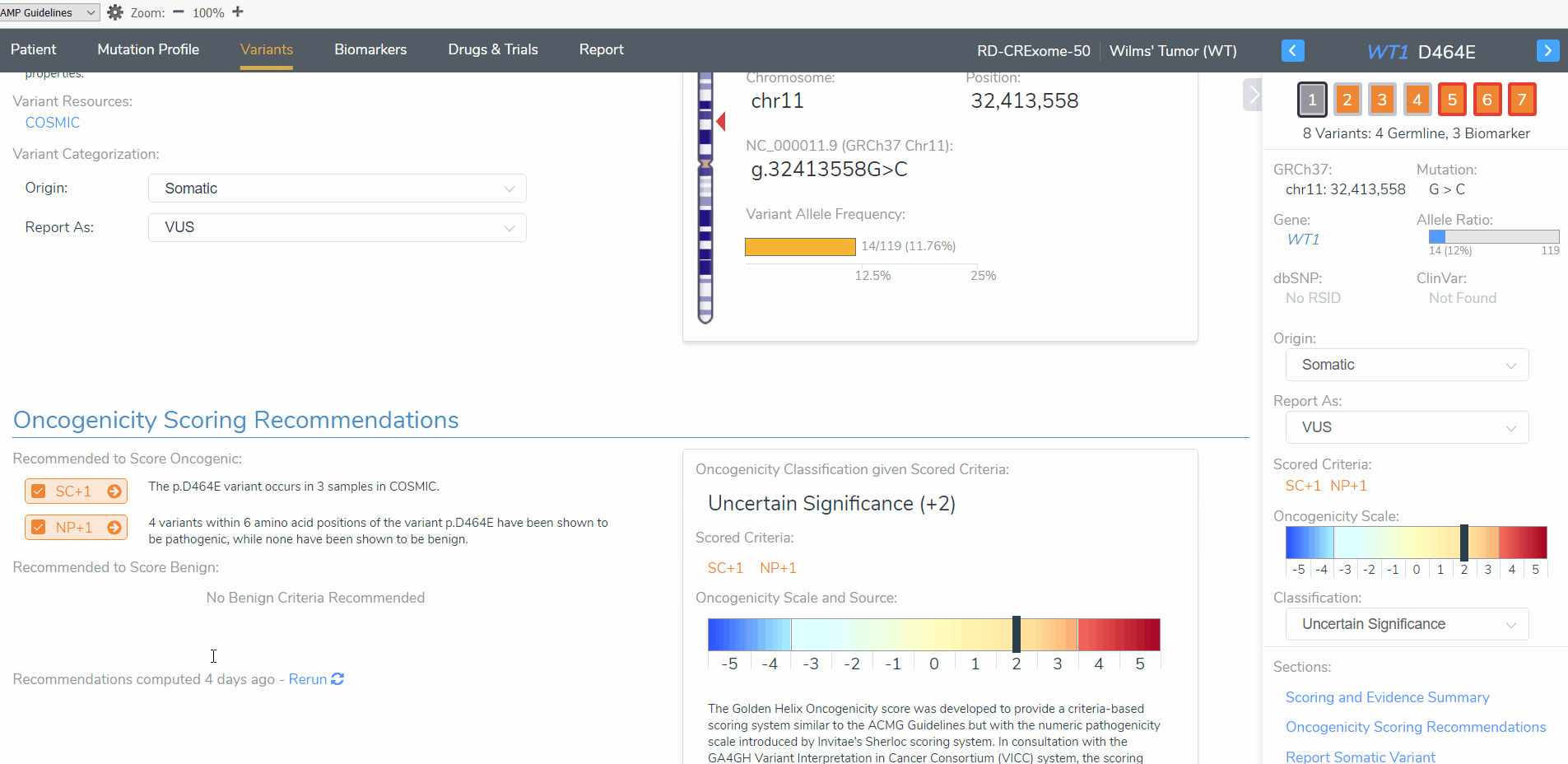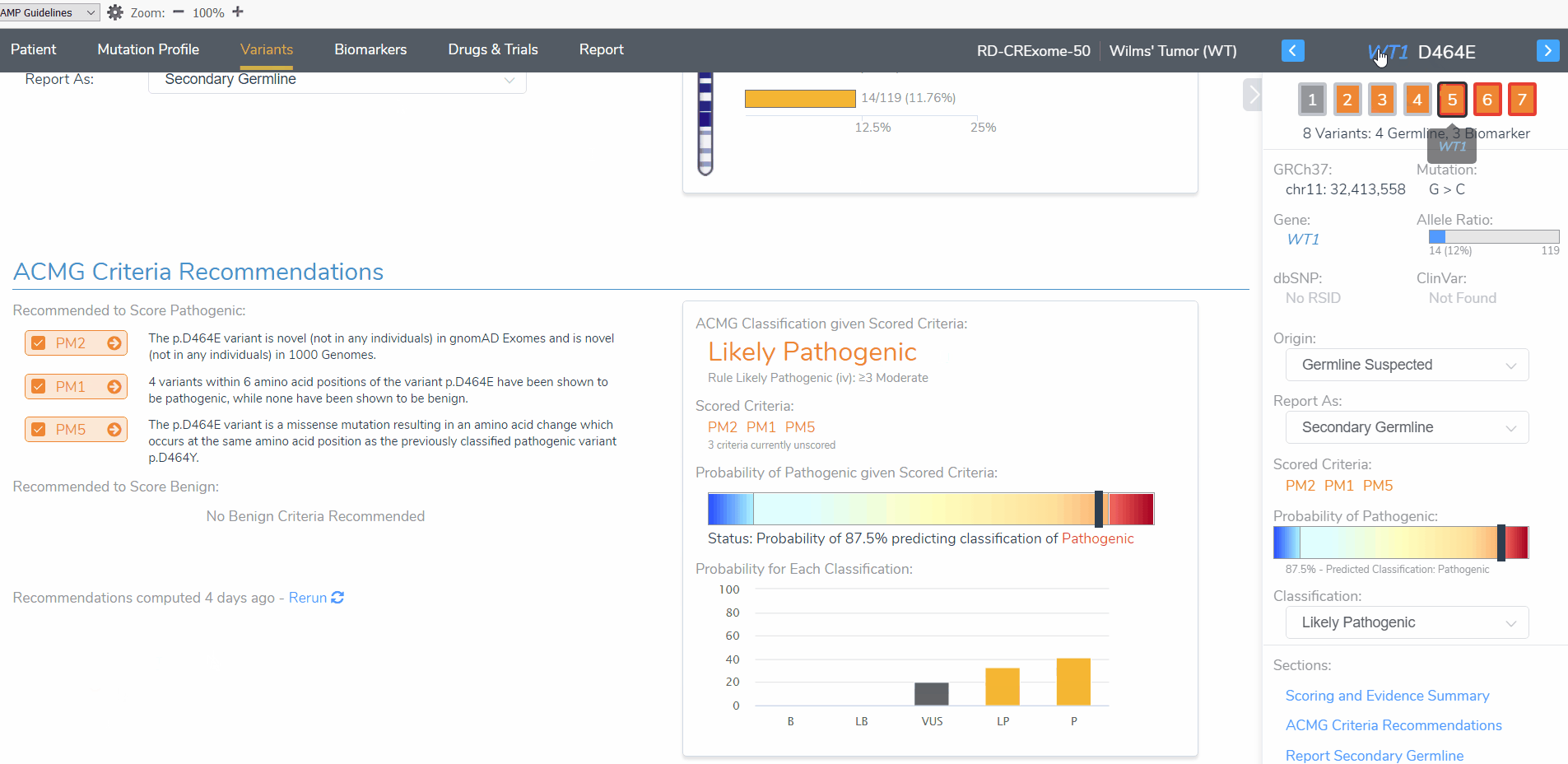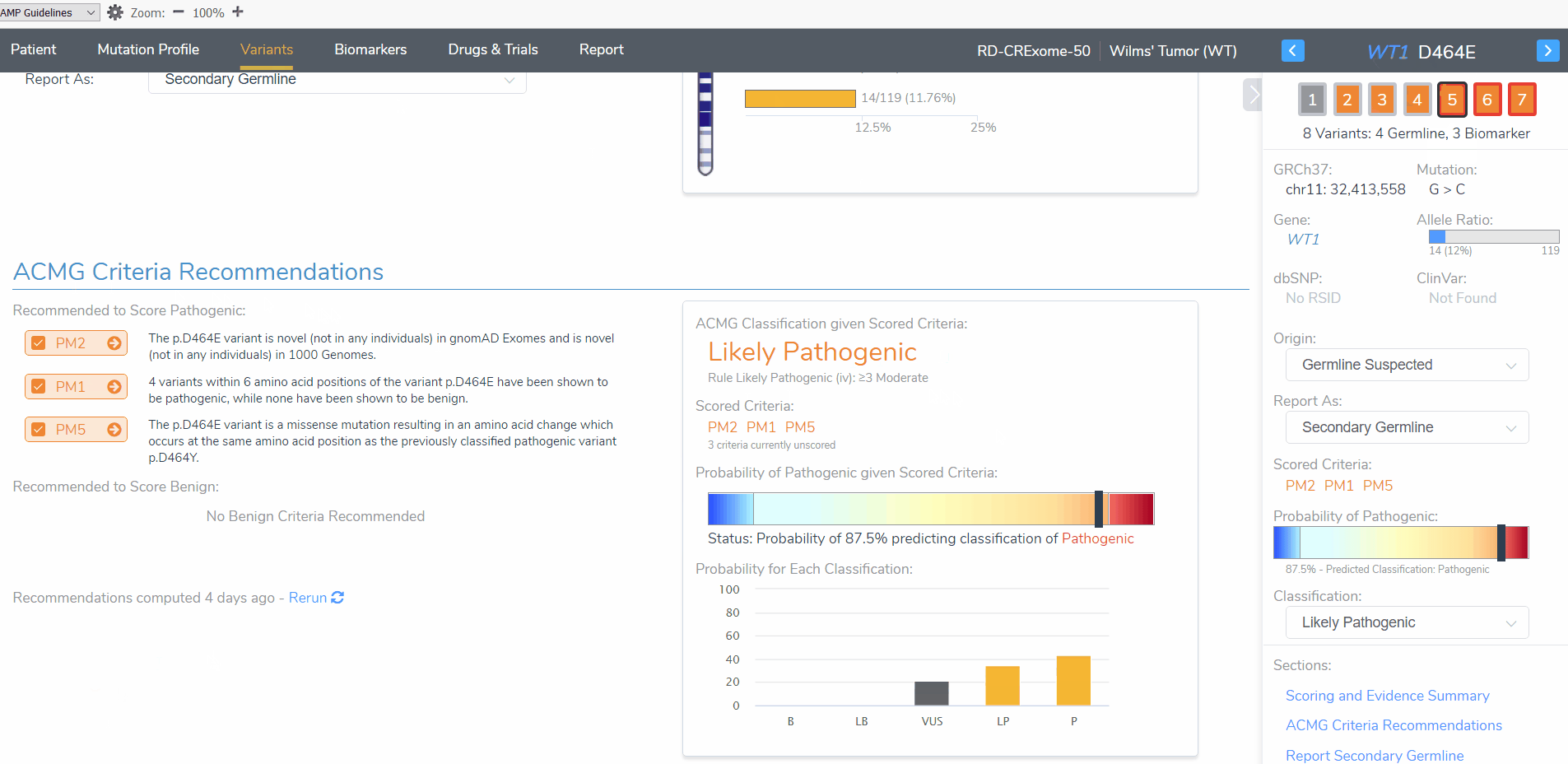
Figure 4: The AMP and ACMG guideline recommended oncogenicity and pathogenicity scoring for the WT1 D464E variant
The WT1 D464E variant is in a mutational hot spot and thus is considered pathogenic/oncogenic according the ACMG/AMP guidelines, respectively. This can be seen even in the other variants that are being evaluated for our patient as these variants are only a few base pairs apart (Figure 3).
The novelty of the WT1 D464E variant according to population catalogs (gnomAD and 1000 Genomes) is recognized by both the AMP and ACMG guideline scoring systems. However, in the case of Mendelian mutations, a rare or novel variant is weighted more in the context of a disease than in somatic mutations. In this case, the WT1 D464E variant was scored +0 on the oncogenicity for being rare or novel in population catalogs according to the AMP guidelines but was awarded a moderate pathogenic score in the ACMG guidelines (Figure 5). Also, even though the D464E variant does occur in the COSMIC database, it is in only in three samples. If this mutation was more common in somatic tumors, the oncogenicity score would have been higher for this variant.

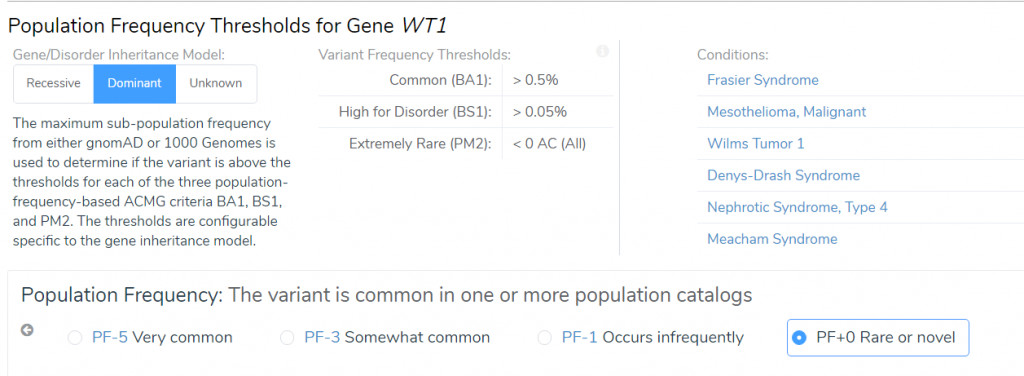
Figure 5: Differences in scoring criteria for the novelty of the WT1 D464E variant
To elaborate on the reasons behind the differences between the pathogenicity/oncogenicity for the same variant, the pathogenicity scoring for germline mutations is usually in the context of a specific disease. Somatic mutation interpretation should be focused on their impact on clinical care, meaning, their ability to predict toxicity, sensitivity, and resistance to drug therapies as well as prognostic, and diagnostic information (5). This helps explain the inclusion of the pathogenic criteria for the WT1 D464 missense variant having an amino acid change at the same position as a previously classified pathogenic variant when evaluating with the ACMG guidelines. The different amino acid may be responsible for a different clinical phenotype than the previously classified variant but is still important when considering the pathogenicity in the context of Wilms’ Tumor. However, this detail has less of an impact on drug therapy and treatment for a somatic Wilms’ Tumor mutation.
In conclusion, no matter if you are evaluating a germline or somatic cancer mutation, VSClinical provides a complete variant interpretation solution that allows users to classify variants according to the ACMG and AMP guidelines in one platform. Germline cancer variants like the WT1 D464 variant discussed in the blog should still be confirmed outside the tumor sample testing and ideally, be compared with a normal sample. Additionally, genetic counseling is recommended to understand the potential impact for the family.
If you analyze somatic or germline variants (or both!), I would highly recommend evaluating VSClinical and testing it with your own data. Here is the link with further details. If you have any questions about this blog or regarding our products, please don’t hesitate to comment below or reach out to [email protected].
If you liked this blog, check out our other blog Using the AMP Guidelines for Rhabdoid Tumor Analysis in VSClinical.
References
1. Andrea Forman and Jilliane Sotelo. (2019). Tumor-Based Genetic Testing and Familial Cancer Risk. Cold Spring Harbor Perspective in Medicine.
2. Maciaszek, J. L., Oak, N., Nichols, K. E. (2020) Recent advances in Wilms’ tumor predisposition. Human Molecular Genetics.
3. National Library of Medicine. 2020, July, 28. Genetic Home Reference. Retrieved from https://ghr.nlm.nih.gov/gene/WT1#normalfunction
4. Diller, L., Ghahremani, M., Morgan, J., Grundy, P., Reeves, C., Breslow, N., Green, D., Neuberg, D., Pelletier, J. and Li, F.P. (1998) Constitutional WT1 mutations in Wilms’ tumor patients. J. Clin. Oncol., 16, 3634–3640
5. Li, M. M, Datto, M., Duncavage, E. J., Wolff, D. J., Younes, A., Nikiforova, M. N. (2017) Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. Journal of Molecular Diagnostics, 19, 4-23



