Golden Helix is excited to release an upcoming VSClinical feature that allows users to analyze next-generation sequencing (NGS) CNV event reporting with ACMG guidelines. This feature will be the first in the NGS workspace to allow this capability and if you are curious about the functionalities you can get a sneak peek by looking at some of our most recent webcasts. Before we begin to shift our attention to these unique capabilities, I wanted to discuss some features within the VS-CNV caller that allow more robust detection of aneuploidy events.

In VarSeq v2.2.1, there was an added feature to subset the considered reference samples to handle extremely large reference sample sets. For example, if there were thousands of references, the algorithm would select the first 100 samples that have a similar mean depth profile to the sample of interest. From there, the algorithm would then compare the coverage profiles over each target interval to identify those that have the highest percent similarity. Those that have the highest similarity would then be selected as the unique reference set for the sample of interest. In some situations, however, this was noticed to have negatively changed the performance of the CNV caller. So, in VarSeq v2.2.2, which has yet to be released, the default number of references will be set to 10,000. This will ultimately maintain the normalization behavior as was seen in VarSeq v2.2.0.
There will also be some changes to the “Advanced” parameters tab in the Target CNV Caller dialog. As seen in Figure 1, there will be new options that include: Signal Scaling for High Percent Difference, Percent of targets to force aneuploidy calls, and Sample Type.
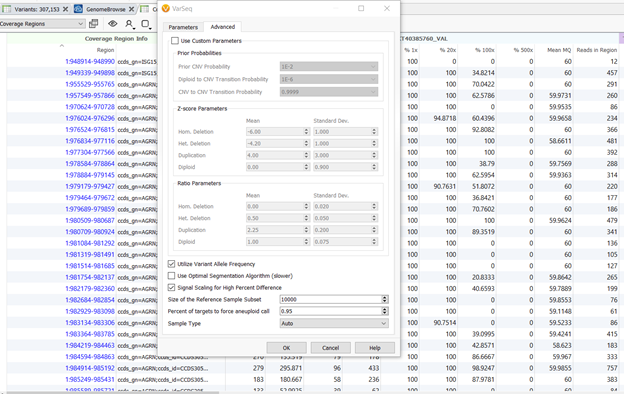
Figure 1. New options in the “Advanced” parameters tab in the Target CNV Caller dialog.
The “Signal Scaling for High Percent Difference” can enable or disable normalization scaling performed on samples with a high percent difference. When there is a high percent difference between the sample of interest and the reference set, there is another level of normalization to help prevent the overcalling of events. In some situations, however, this feature was hiding validated events in non-autosomal regions and thus deselecting this feature allowed for proper detection.
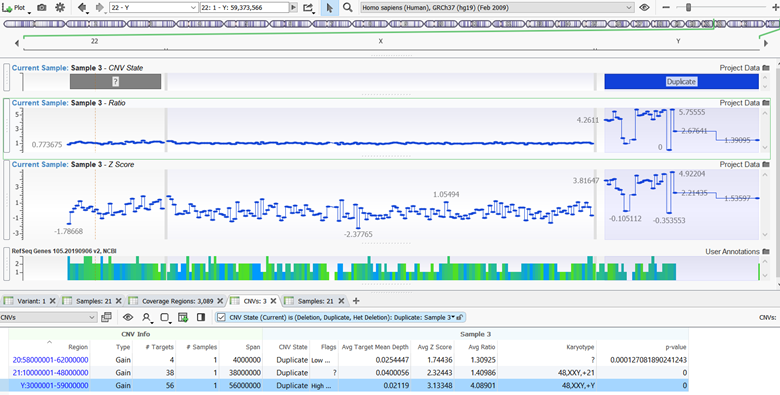
Figure 2: Signal Scaling for High Percent Difference can be deselected to prevent compression of noisy regions.
The other new parameter will be the “Percent of Targets to Force Aneuploid Call”. This feature will force an event to be called a whole chromosome event if the percentage of targets in a given state exceed the selected percentage, which is 95% by default. This ultimately takes the collection of all CNVs in a chromosome and modifies the minimum number of events needed to call the entire aneuploid event. In other words, if the percentage of deleted or duplicated targets in a chromosome exceed the selected threshold, then the entire chromosome will be called as an aneuploid deletion or duplication event.
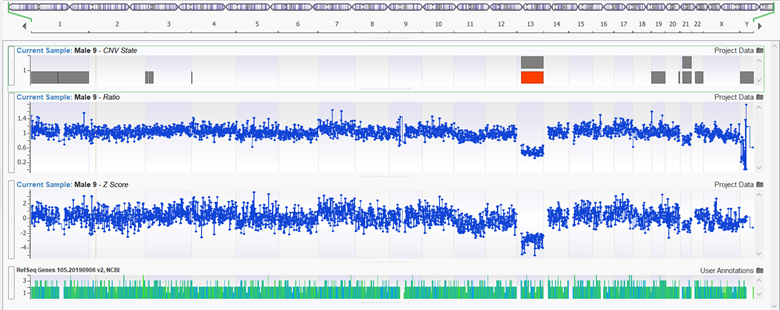
Figure 3: Percent of targets to force aneuploid calls can be lowered to capture whole chromosome events more effectively.
Lastly is the new parameter “Sample Type” which can be set to either Auto, Gene Panel, or Exome for inferring the sample type. If the sample type is exome, the algorithm is permitted to call whole chromosome events and report these events in the Karyotype output at the sample level. With this feature, the user may force this option if you desire to call whole chromosome aneuploidy events, especially in the X or Y chromosomes.

Figure 4: Select the Sample type to be Exome will lead to better representation in the Karyotype output results.
I hope you enjoyed reading this blog and are excited about the upcoming release of our VSClinical ACMG CNV classifier. The goal of this blog was to begin to introduce more information about our CNV event reporting with ACMG guidelines. Stay tuned for more updates on this and don’t forget to register for the upcoming ACMG CNV webcast on November 4th. As always, if you have any questions about our software or the content described above, feel free to contact [email protected].



