VarSeq recently received major upgrades in a wide range of areas, one of these areas includes adding annotations such as GnomAD. This includes new fundamental methods of CNV ACMG guideline processing but also a large number of small additions in annotations. One addition is the application of gnomADs – Gene Constraints. This provides various metrics for pathogenicity on a per gene or transcript level. Figure 1 shows the list of fields contained in the Gene Constraint track.
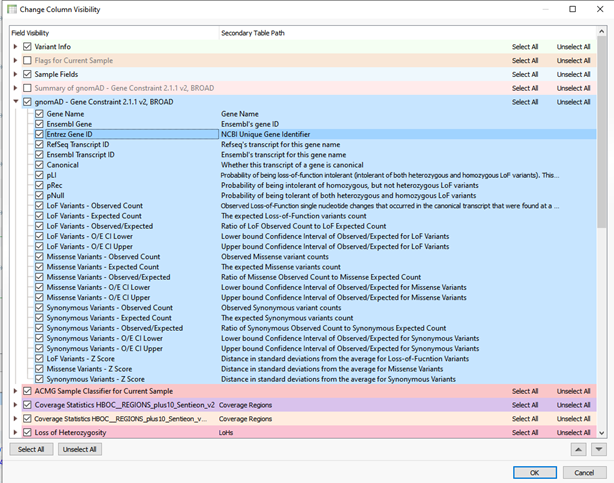
Aside from breakdown of different variant types with their observed vs expected counts, confidence intervals, and zscores, here are some key metrics present in gene constraints:
pLI: Probability of being loss-of-function intolerant (intolerant of both heterozygous and homozygous LoF variants). This metric estimates the probability that a gene falls into the class of LoF-haploinsufficient genes
pRec: Probability of being intolerant of homozygous, but not heterozygous LoF variants
pNull: Probability of being tolerant of both heterozygous and homozygous LoF variants
This information is interesting to explore in the variant table itself, especially when working through gene discovery for common and rare diseases. However, these key fields are presented in VSClinical. When reviewing any individual variant’s impact on a gene, there is a section dedicated to the gene’s tolerance of various mutation types (Figure 2).

This section not only lists the pLI, Z-score, and Obs/Exp LoF frequencies but also defines the final status (example: Intolerant of LoF variants) for this variant assessment in the MSH2 gene. Additionally, you’ll find a link to the gnomAD blog for more details. This track automatically is incorporated into the VSClinical variant evaluation tool.
I hope you enjoyed reading this blog post detailing GnomAD. Please reach out to us at [email protected] if you would like some training, or would like to explore the database in more detail. Feel free to also check out some of our other blogs that always contain important news and updates for the next-gen sequencing community.




Hello, thank you for the very useful blog. I wonder whether there is any metrics that can suggests mechanism of pathogenicity (loss of function vs gain of function) please. Thank you
Yes! We have a number of annotations and predictive algorithms for exactly that purpose. The main one would be our ACMG autoclassifier for pathogenicity. We can also leverage databases like ClinVar to look for records of pathogenic variants, or use RefSeq to look for LOF or missense variants. Send a request to [email protected] if you would like to talk to one of our FAS about your specific use case.