Up until a few weeks ago, I thought variant classification was basically a solved problem. I mean, how hard can it be? We look at variants all the time and say things like, “Well that one is probably not too detrimental since it’s a 3 base insertion, but this frameshift is worth looking into.” What we fail to recognize is just how many assumptions went into the above statement. What transcript set are we using? In what part of the gene did the mutation occur? What subfeature of the gene are we looking at? Are there other ontologies for the variant? Why did we use the term we did? In order to develop a tool for variant annotation, rules to answer all these questions have to be codified into the software. Enumerating these assumptions means that a process that is subject to a great deal of human interpretation, is now a rigidly defined objective framework. There are currently three major tools that attempt to classify variants: Annovar, SnpEff and Variant Effect Predictor (VEP). It is no surprise that these tools do not always agree since the way the rules have been defined differ slightly between each application.
In a recent paper, Davis McCarthy et al. showed that both the choice of annotation software and transcript set can have a significant impact on the classification of variants. McCarthy compared Annovar and VEP. He also compared running Annovar using both the set of transcripts available from Ensembl and the commonly used set of RefSeq transcripts curated by NCBI. The paper’s key conclusion was that there was only about 65% concordance between the annotations for Loss of Function (LoF) variants produced by the two tools using the same transcript set. Using different transcript sets, the paper reported only 44% concordance among putative variants using Annovar.
I was so surprised by the finding I wanted to get a better feeling for the type of variants that cause these tools to differ. I also wanted to see how the widely used application, SnpEff, stacked up to the other two tools.
Since I was most interested in trying to find the types of variants that caused discrepancies between annotation algorithms, I decided the best way to dig into the problem was to create an artificial set of variants in the widely studied CFTR gene. Using Python I generated a set of variants amongst all positions covered by an exon in Ensembl’s database with 100 bp margins on either side (IPython Notebook). At each position I created:
-
All possible SNPs
-
All possible 1 base insertions following that position
-
Two possible 2 base insertions following that position
-
Two possible 3 base insertions following that position
-
Each possible 1, 2 and 3 base deletions following that position
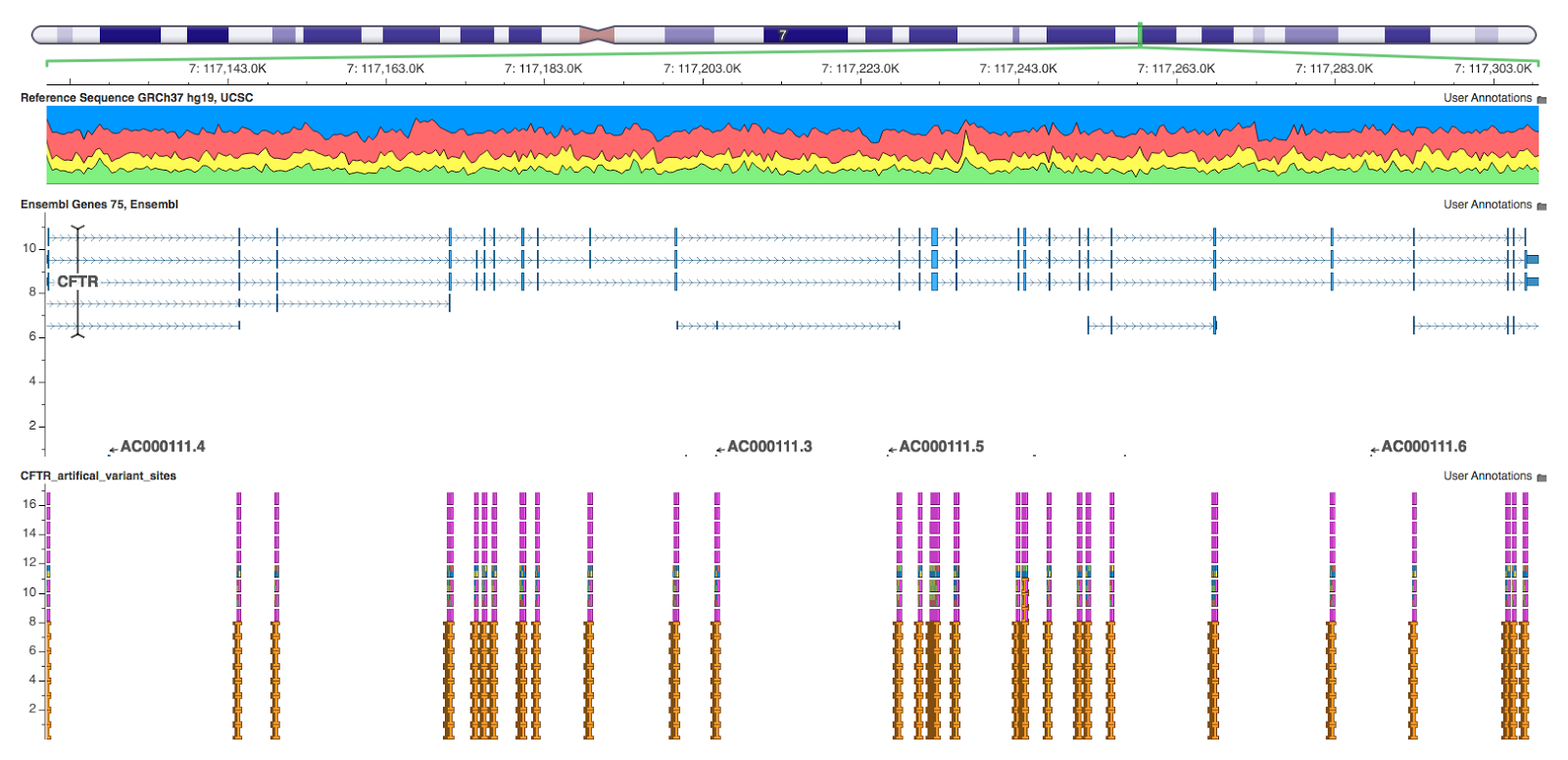
I then output this variant set to a VCF file and ran it through VEP, SnpEff, and Annovar using both Ensembl and RefSeq transcript sets (IPython Notebook showing VCF parsing).
At this point, a degree of subjectivity must enter into the analysis. I quickly had to start making decisions about how to compare annotations between tools. I did this comparison in Python using Pandas and HDF5 to back the analysis (IPython Notebooks normalizing annotations and analyzing Ensembl and Refseq comparisons).
The State of Variant Classification
The state of the variant annotator is…hmmm…complex? It’s extremely fragmented with each tool choosing to represent its results differently. But, the differences are not just superficial. Each algorithm makes significant choices about how to label variants. As mentioned above, these don’t always agree. Sometimes the tools differ in ways that are both arguably correct, but in a few cases, the generated annotations are flat out wrong.
Performing a perfect comparison of the three tools is frankly impossible. To start with each tool outputs its annotations in a different format. Annovar’s output is a tab separated file, while SnpEff and VEP produce VCF files which use the “INFO” field to encode their annotations. While SnpEff and VEP represent data in a consistent format, the format of Annovar’s rows changes depending on context. For example, Annovar uses the gene field to provide distance information for all intergenic variants. Similarly, for variants labeled as splicing, it overloads the gene field with HGVS notion. These decisions make sense from a human point of view but can cause problems when parsing the file. Since it is common to annotate thousands if not millions of variants at one time, I’d argue that the ability to easily parse the output file should be paramount when designing the tool.
Each tool uses a different nomenclature for defining sequence ontology. These differences encompass both semantically equivalent terms and terms which are similar but vary in their specificity. To compensate, I was forced to make some subjective decisions about how to normalize terms. Ultimately, this meant bucketing terms together. Since Annovar’s annotation categories are the most general, I used these to build my categories.
There is some hope that variant annotation in the future will start to use the standard ontology terms defined by The Sequence Ontology Project. Even with standardized terms, it is still extremely important to use precise language when classifying variants to avoid ambiguity during the discussion. For example, all the tools have a concept of a splicing variant. Using a term like splice_region_variant doesn’t really clarify the impact of the variant. The Sequence Ontology Project uses splice_region_variant as a parent category (an inner node in the sequence ontology tree). Thus, using this term all we’ve communicated is that the variant could be up to 3 bases exonic or 8 bases intronic of a splice site. The variant’s location within this interval could drastically alter the impact. When we actually want to communicate the punitive effect it is more useful to use a specific term like five_prime_cis_splice_site.
Finally, each piece of software deals with a single genomic variant producing annotations against multiple transcripts in a different way. If a gene contains more than one transcript, a variant will have multiple annotations depending on where it is located with respect to each transcript. Moreover, some genes overlap so a single variant could affect multiple transcripts on multiple genes. Both SnpEff and VEP list each way the variant was classified. Annovar instead returns only the most deleterious variant based upon a priority system. By necessity, I tried to follow this prioritization in order to compare “apples to apples”.
Although it might seem reasonable to collapse variants down to single most punitive annotation, doing so poses a multitude of problems. First, it imposes a subjective determination of what is the worst type of variant. Is a frameshift really worse than a stop gain? Maybe, maybe not. A frameshift might occur early in a coding sequence, but might be quickly offset by another frame shift. This situation is likely to arise since many variant callers (e.g. GATK’s UnifiedGenotyper) prefer to call small allelic primitives. However, it might be reasonable to argue that it makes little difference whether an indel is a frameshift or stop gain since they are both Loss of Function variants.
Second, collapsing annotations removes granularity that can be useful while filtering variants. Perhaps, your protocol only calls for looking at stop gain variants. If the classifications have been collapsed by the software, you are out of luck. I predict this collapsing of lower priority variants will become a larger issue in the future as the impact of variants outside of coding regions is discovered to be significant. In the future, a SNP in certain regulatory regions might be found to have a greater effect than an inframe substitution in a coding region.
Another interesting decision Annovar made is not to annotate variants that lead to start loss/gain mutations. While I can see the reasoning behind calling a start gain just a nonsynonymous variant, not annotating start losses is troubling. A start loss potentially results in the complete loss of a transcript’s product. In fact, SnpEff predicts the effect of start losses as high impact.
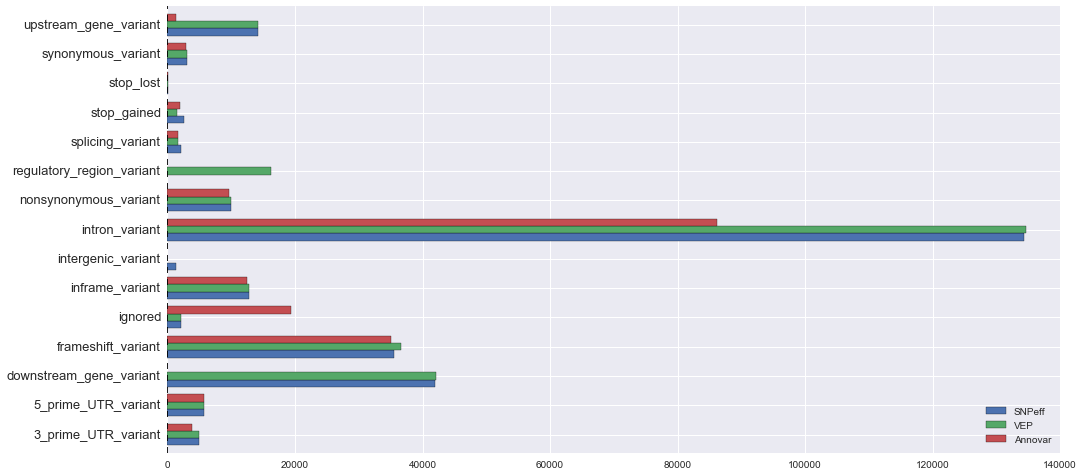
The concordance between the three algorithms in coding regions is relatively good. When we exclude variants with annotations that imply a non-coding region (ie downstream, upstream, intergenic, intron, 3 prime and 5 prime) and annotations in the “ignored” category there is 93% concordance between the algorithms. However, when these categories are included the concordance falls to 49%.
While this second number seems shocking, it’s less concerning as it is depressed by annotations that are similar, but vary in levels of precision. Additionally, if even one algorithm disagrees, it cannot be counted as a match.
Most of this variation in annotations is explained by the way each tool defines non-coding features. For example, SnpEff, uses 5kb to define upstream and downstream regions, while Annovar uses 1 kb. In many other cases, variants in non-coding regions were bucketed into the “ignored” category.
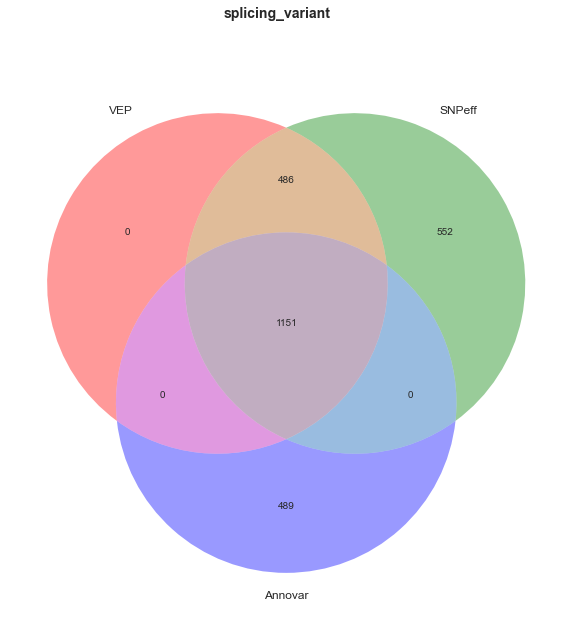
Next, note that only VEP annotated regulatory region variants. Since I used the web interface to VEP, this was done without having to hunt down additional datasets and get them into a format that the tool could use. This feature is a nice bonus for VEP users.These variants would likely be annotated as intronic or intergenic by the other algorithms decreasing non-coding concordance.
Another distinction for VEP web users to note is the available sets of transcripts. Ensembl is of course available, but the RefSeq database also contains CCDS and Expressed Sequence Tags. This caused the RefSeq comparison in my analysis to not be reliable; hence I focused on annotations using the Ensembl transcript set.
Given that the coding concordance is relatively good, but not perfect, we can say that we have a small but significant amount of disagreement. For example, in the categories of splicing, frameshift, and stop gain there is a non-trivial amount of disagreement. It is in these dark edge cases, where the demons of variant annotation hide.
Specific Variant Test Cases
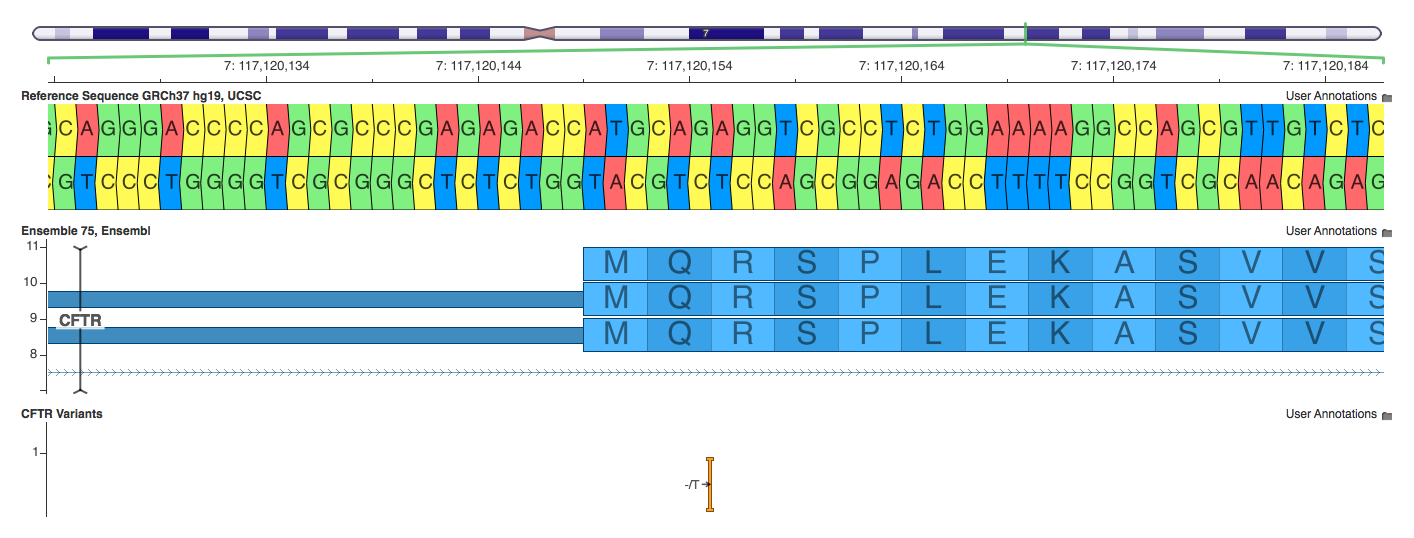
7:117120154-117120154 -/T
Annovar and VEP classify this variant as a frameshift, while SnpEff classifies it as stop gain. Who’s right? Tough to say. It is a frameshift, but calling it a stop gain is more precise.
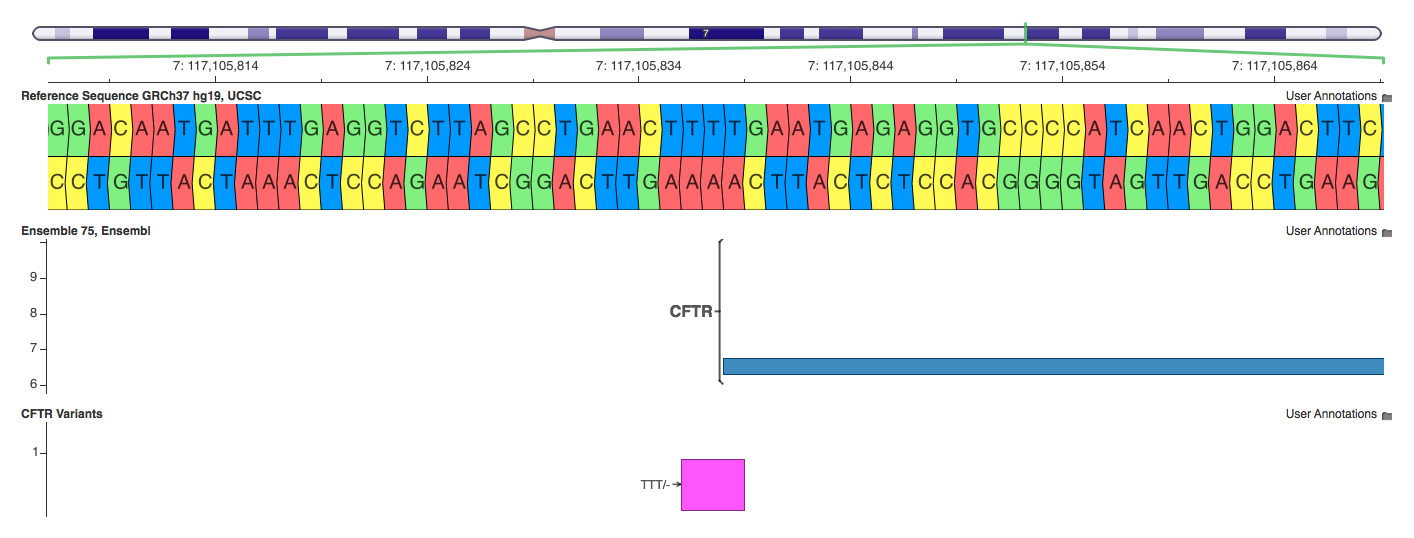
7: 117105836-117105838 TTT/-
What is interesting about this annotation is that VEP is looking at every base affected by the indel. Thus it figures out that the T at 117105838 is the first base of this CFTR Exon and annotates the variant as a non-coding-exon variant, whereas Annovar calls it intergenic and SnpEff calls it an Exon, intergenic and upstream variant. This is a good example of SnpEff annotating all possibilities, even if two of the possibilities are mutually exclusive (exon and intergenic).
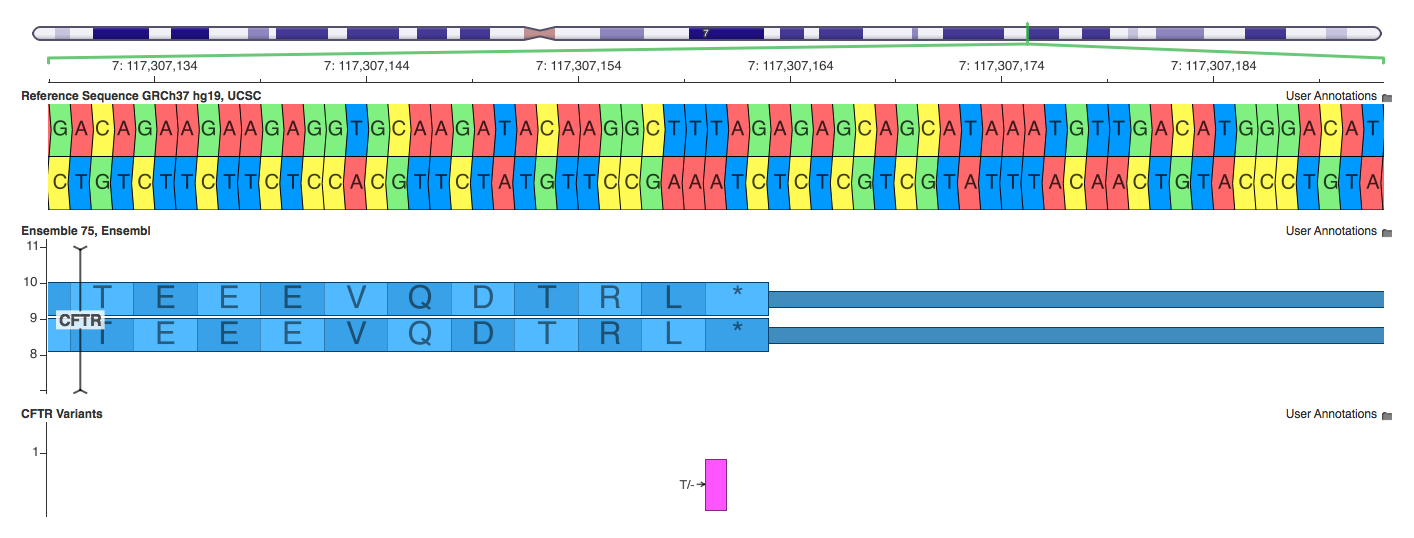
7: 117307160-117307161 T/-
Both Annovar and VEP correctly label this variant as a stop loss. SnpEff labels it as a frameshift, which is true, but less precise.
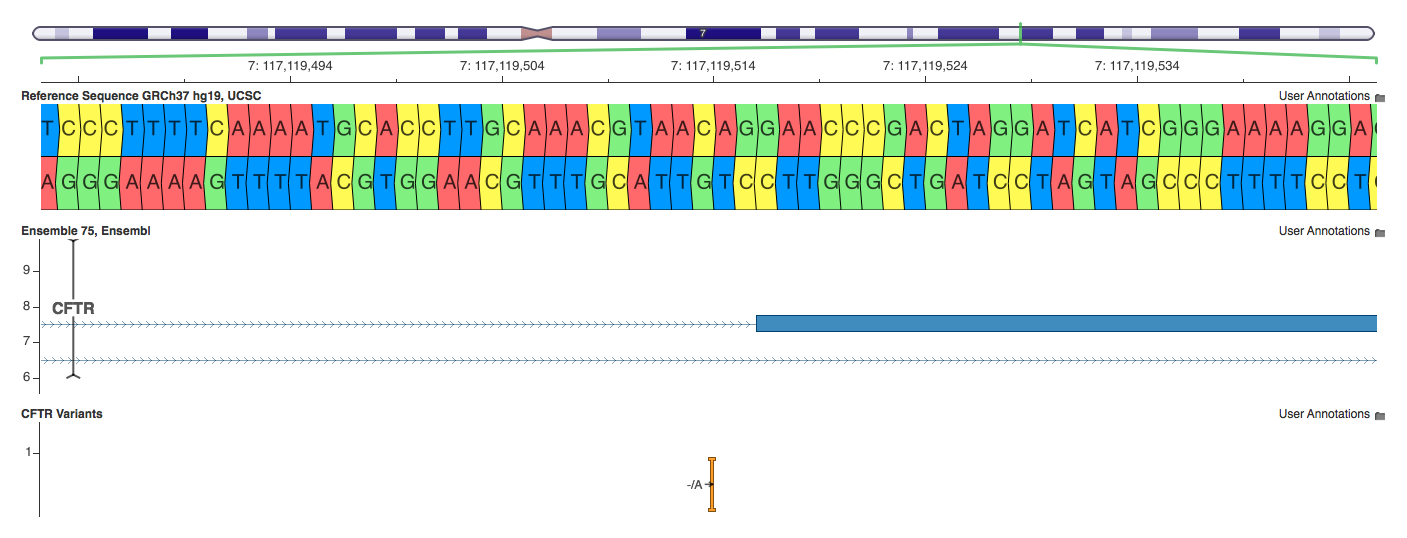
7:117119513 -/A
This is an interesting and subtle case which elucidates why splicing variants displayed a lower amount of concordance. SnpEff annotates this as a splice_site_acceptor — technically wrong since it is 3 bases intronic. VEP annotates it as a splice_site_region — correct. Annovar annotates it as intron — true, but imprecise. The key takeaway is that all three are using different terms to describe the same variant, making comparative analysis extremely difficult.
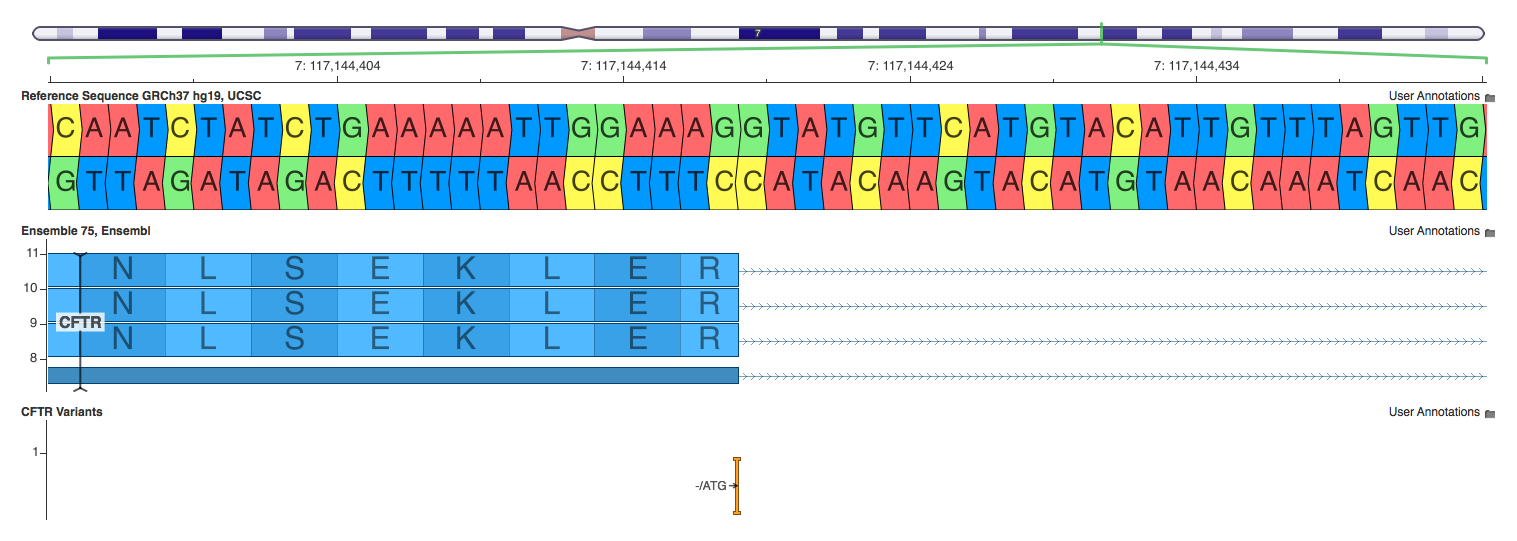
7:117144417 -/ATG
One discrepancy that is worth investigating is the case of a stop codon being inserted after the last codon of an exon. VEP and Annovar call this a stop gain, while SnpEff calls it a splice site variant. If we think about how the transcription process works, we can infer that this variant would act as a stop gain. The first two intronic bases (GT) act as a signal for the intron to be spliced out. However, the stop codon is inserted just before these two bases. Thus, during translation, the ribosome will hit the stop codon and stop producing the protein rather than splicing the two exons together.
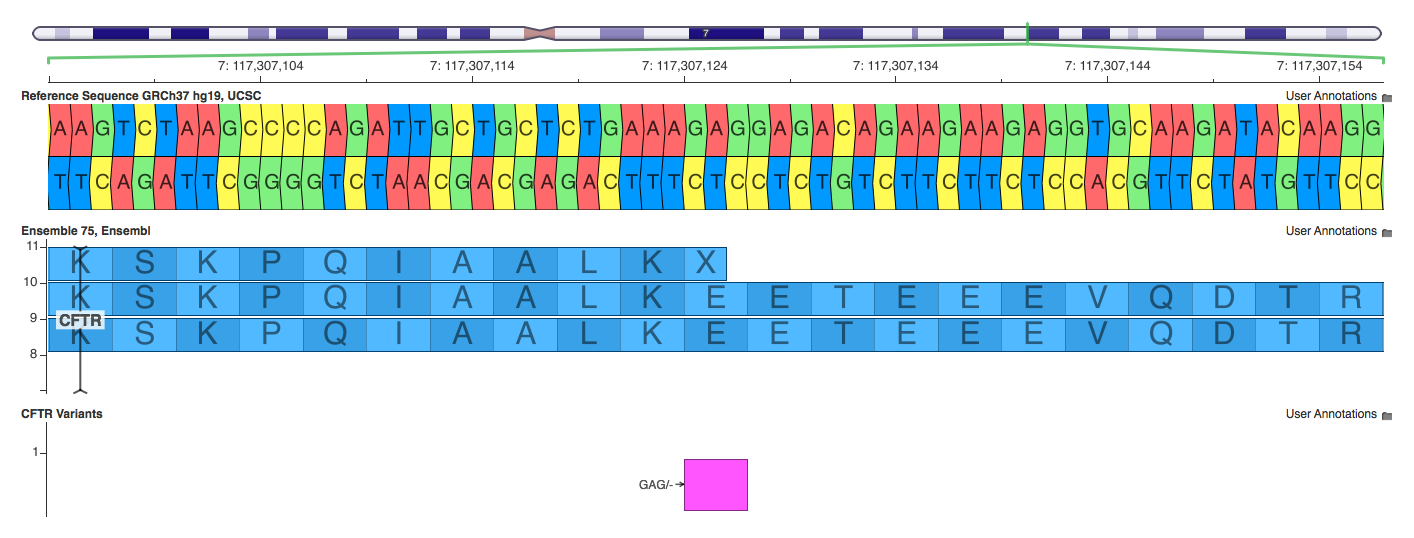
7: 117307124 – 117307126 GAG/-
This 3 base pair deletion is a great example of a variant that causes the algorithms to use substantially different annotations. SnpEff labels it a frameshift mutation. This classification is wrong with respect to any transcript at this position. SnpEff makes this determination using the incomplete transcript (the one on top in the screenshot). VEP accurately calls it an 3_prime_UTR_variant against the incomplete transcript and an inframe deletion against the others. Annovar correctly calls it a “nonframeshift deletion” and uses the bottom transcript to make that determination.
Pulling on a Thread: Examining Transcript Sets
I wish all we needed to worry about was how different tools annotate variants. As discussed above some of the classifications disagree, but often filtering is performed on an aggregate class of variants (i.e. coding or loss of function). In these cases classifying a variant as a frameshift instead of a stop gain will not change the set of resulting variants. Even where a classification is flat out wrong, it is easily verifiable by viewing the variant in a genome browser. Additionally, with the availability of multiple variant classification algorithms, the ability to use another tool exists. Most importantly, we can talk very precisely about the variant since we assume the coordinates and transcript mappings to be accurate.
But, can we actually make this assumption that the transcripts we are using have been accurately mapped to the genome? As it turns out this mapping is the more confounding problem when classifying variants.
Transcript sets are are available from different organizations such as Ensembl and RefSeq. To understand what is included in these databases it helps to understand the origin of the data. Transcripts are usually determined by sequencing RNA or determining the primary amino acid of proteins, although some transcripts are computationally inferred. From this data, we can build up small pieces of the transcriptome. Finally, to place these pieces in genomic space we need to align these sequences to the reference sequence.
Before discussing the specific issues that affect transcript sets, it’s worth thinking about why it is such a problem. Having an accurate transcript mapping is crucial because it is used quite far upstream in the NGS data analysis pipeline. If the transcript is not placed accurately on the reference genome then all downstream analysis using this derived data will be inaccurate. Moreover, these inaccurate mappings are more difficult to spot than a misclassified variant. Finally, it makes it much harder to speak precisely about a variant since it’s context may be incorrect. Essentially, if we are unable to rely on transcript mapping, we have added a wide plane of uncertainty to our analysis.
When we look at a VCF file full of variants, we are seeing their positions in genomic coordinates — that is, we know their positions with respect to the reference sequence. However, when examining a disease causing variant, the variant’s genomic position is of secondary importance. What we care about is its biological impact. This functional impact is much more tightly linked to their position in coding space (i.e. HGVS c dot notation). Thus, we need to either translate the variant into coding coordinates or translate the transcripts’ coordinates into genomic coordinates. Unfortunately, it is impossible to translate a small variant directly into coding coordinates since there are a multitude of possible positions to which it could map. Thus, we are forced to map transcripts back to the genomic reference sequence.
This use of genomic coordinates implicitly assumes that there is such a thing as a canonical reference sequence. Although the human reference has come a long way from it’s first release, it still has many gaps, tiling issues and alleles that are rare or non-existent in humans. The alignment algorithms used to mapped transcripts back to genomic space must contend with these issues in addition to novel challenges such as the expectation of large gaps (introns) in the alignment. When mapping transcript sets such as RefSeq to the reference, these issues manifest themselves in the creation of exons and introns that are not in their correct position or break the biological rules of translation. Needless to say, the problem is difficult to solve and thus leads to imperfect alignment and ab initio gene construction.
At the recent Human Variome Project meeting in Paris, Invitae’s Reece Hart, gave an extremely important talk about just what a significant problem this mismapping is. He was able to show that the mapping for nearly 3% of transcripts in the NCBI RefSeq database that UCSC has aligned to the reference the coordinates is likely inaccurate.
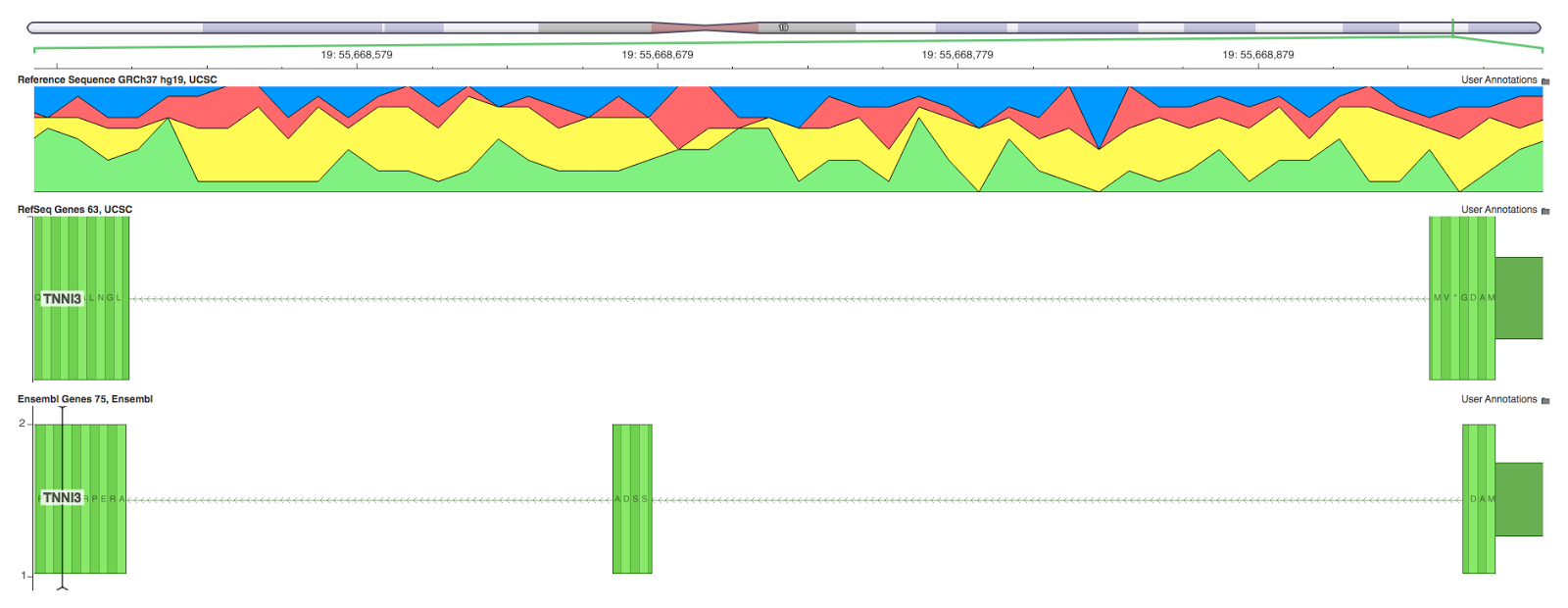
NCBI reports the transcript RNA sequence, but doesn’t provide genomic coordinates. [As Deanna pointed out below, NCBI does provide the genomic alignments]. UCSC, however, uses the RNA sequence provided by the NCBI RefSeq project and aligns it to the reference using the BLAT aligner. In some spots, BLAT makes a mistake and it is clear that the transcript mapping is biologically impossible. For example in the TNNI3, we see that BLAT doesn’t split exon 1 into two introducing the appearance of a premature stop codon 4 amino acids into the protein. Examining Ensembl’s mapping of the same transcript, we see that the Glycine codon is properly split preventing the stop codon. But it gets worse! BLAT uses an extra nucleotide in it’s alignment (UCSC reports the coding sequence length is 841 vs. the 840 that Ensembl reports). This causes the gene mapping to be frame-shifted 24 bases into the coding sequence! Thus, if your variant occurs anywhere in the remaining 816 bases it will be annotated incorrectly even by a perfect algorithm.
TNNI3 is an extreme case and was easy to spot because of the premature stop codon. Some of the other cases are not so obvious. For example, BLAT moves exon 12 in CARD9 323 bases and changes it’s length from 7 bases to 5, however, the rest of the sequence is left intact so this would only affect a variant that was in exon 12, but the likelihood of discovering this without prior knowledge is slim. Another interesting case is the EMG1 gene that I tweeted about last week.

So is the solution just to use the Ensembl transcript set? Regardless of how you answer this question, we don’t live in an ENST world and the dominance of “NM_” nomenclature means that we still need to provide RefSeq names. Furthermore, Ensembl is trying to produce a comprehensive set of transcripts for each gene, not a high confidence list of well studied transcripts. This breadth adds a lot of “noise” to an analysis pipeline. Thus, the large Ensembl transcript set might be suitable for a research environment but is less suitable in a clinical bioinformatics, gene panel or exome analysis pipeline.
One devilish example of Ensembl’s “noise” is that transcripts with incomplete coding sequences are included. Often this causes the transcript to start out of frame. The GTF file spec handles this with a frame field. However, if a tool doesn’t handle this out of frame start, inaccurate annotations can be produced. I believe this is what happened with the classification discrepency of a single base deletion (7:117267851 -117267852 T/-). On transcript ENST00000468795, SnpEff called this a frameshift, while VEP correctly called it a stop lost. If the transcript was not adjusted it’s easy to see why SnpEff would miss it. Thus, this is a case where having a more complete data set introduced complexities into the underlying data that the tool (or data curator) must account for.

The effect of including all detected transcripts can cause additional edge cases that the data curator must be aware of. Here an incomplete CDS in Ensembl’s transcript set for CFTR starts out of frame. The top transcript is properly adjusted for the reading frame and thus ends in a stop codon. The bottom transcript ignores the frame parameter, resulting in the incorrect representation of all codons downstream of the exon which began out of frame. Without accounting for this irregularity, downstream analysis can become inaccurate.
One recent attempt to improve the accuracy and usefulness of variant annotations is the Loss of Function Transcript Effect Estimator (LOFTEE). This just-released software package is written by Konrad Karczewski and fits on top of VEP. It removes variants within a short distance (less than 15bp) intronic regions, non canonical (ie not GT AT signature) splice regions, LoF variants in the last 5% of the transcript, and variants where the LoF allele is the ancestral allele for that position. Each of these decisions can help to improve our confidence that the variant we are looking at is punitive.
Ultimately, we have to realize that there are many factors that enter into variant annotation and genomic interpretation, and that it may not be in the class of problems that has a single correct solution. Instead, it may be the type of problem with many acceptable solutions whose value varies with context. I think the most important thing that you can do is to familiarize yourself with the transcript set you choose to use — know the limitations of RefSeq mapped by UCSC and how Ensembl can obfuscate results. You could consider using CCDS too. This database represents the intersection of the set of transcripts in Ensembl, RefSeq, and HAVANA. Since it only represents coding transcripts, it removes some of the less studied transcripts found in the comprehensive Ensembl. But, don’t stop with the transcript set! You should become very familiar with your classifier — study it’s documentation, run some test variants through it and study them in a genome browser. Also, think about what really matters in your analysis — can you bucket annotations in terms of impactfulness or do you need to know the specific type of variant.
If you made it this far, I want to offer my congratulations! I took an in depth look at variant classifiers over the last few weeks because I am building a new one and want to hear your feedback. What’s important in terms of annotations? How do you curate your data? How does it integrate into your current pipeline? We want to hear from you and incorporate the features you need into our next generation of the variant classifier.




“NCBI reports the transcript RNA sequence, but doesn’t provide genomic coordinates.”
This is not true. NCBI provides a GFF3 file of all gene annotation per build.
ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens/GFF/
You can also get this information from Entrez Gene, though it is a little harder.
Thanks for pointing this out! We’ve been looking for this exact type of data for years! It will make data curation much easier.
I just took a quick look at it in GenomeBrowse and it was good to see that many of the alignment issues present in the UCSC aligned transcripts (e.g. in the TNNI3 and EMG1 genes) are not present in this NCBI source. I think it just emphasizes how important it is to be aware of the upstream that is being used during annotation since we now know that not all data labeled “RefSeq” is the same.
A while back I ran into similar problems when I wanted to understand differences in variant annotations between VEP and our own tool, CooVar: http://www.biomedcentral.com/1756-0500/5/615
It started as a seemingly simple exercise and turned out to be quite an effort, with some VEP annotations remaining unexplained.
Get in touch with us if you’re still struggling with the annotations and we can see what’s going on. We’re [email protected].
Very true. I thought the comparison would be much simpler and that I would be able to more easily match categories in a 1:1 fashion. Part of the issue is due to Annovar, having a very different approach to delivering data to the user — that is, collapsing annotations based on a prioritization scheme. In any case, I came away with the sense that within coding regions, all the algorithms seem to do okay with the easy annotations, it’s just the more funky ones where they disagree.
CooVar looks really interesting! I like that it just takes the raw GTF as input. Part of the reason why I just used the provided transcript sets for my comparison was that I didn’t want to do a custom curation for each tool.
Very interesting post and highlights several issues that I’ve had to get across to collaborators when they complain about some abiguities in data.
Conflict of interest: I am a very happy VEP and ensembl user, so I may be biased in their direction.
A few points though. VEP can return tab-delimitted results thereby giving a consequence for each variant in each transcript on a separate line. This, in my opinion, is the only way to do variant calling. Prioritising consequences or using canonical transcripts only is wrong and should never be used in any context.
You mention that tools should use the sequence ontology. I agree and VEP does.
Finally, you say we live in a ‘NM_ dominated’ world. Firstly, that’s a very north american centric view. Secondly, it’s not true. We live in a very gene-centric world (where the central dogma of one gene to one protein is still entrenched), so getting collaborators to think in a transcript-centric way is a challenge especially as you get closer to the clinic. They want to know which gene is affected, but as we know transcript isoforms are often tissue specific, so the answer is sometimes ‘it depends’. Not very satisfactory 🙂
The computer scientist in me really likes the way Ensembl organizes their data — clean, unique identifiers, using standardized terms, easy to parse files etc. VEP’s VCF format is very easy to work with. It contains all the data in the places you expect, so it essentially came down to a couple extra splits on commas and pipes and then restacking the data in pandas. In a pipeline, the tsv would probably be a better choice since these operations do take a bit of compute time.
You hit the nail on the head that RefSeq identifiers are widely used in North American institutions. Our experience is that the closer you get to the (North American) clinical space the more true this becomes (or they start using HUGO gene names). Thus, I agree with you on Gene-centric point. We often hear from the clinics that they really think in terms of a person being positive or negative at the gene level. However, this whole mapping/collapsing from that variant level to gene level introduces even less precision into the conversation. Not very satisfactory, indeed!
Really great blog and useful feedback for us at Ensembl. I’ve sent this to our VEP developers so they can see if there’s anything we can improve.
One thing that you’ll see in release 76 of Ensembl (next month!) is GENCODE basic as a transcript set for human and mouse in the VEP. This is a pared down transcript list that removes incomplete transcripts (except where the gene only has incompletes), which will solve many of the problems you encounter in the second section.
Ooh. That sounds like a great idea. I’ll look forward to it.
This sounds great!
I actually looked around for a way to pull GENCODE Basic into a transcript set that we could use internally and there wasn’t a straightforward way to curate the data, so I’m really looking forward to the release. I think GENCODE Basic really hits the sweet spot in terms of information that will cover 90% of people’s desired use cases.
None of the pictures (past the signpost photo) in the article are displayed here. When opening the pictures’ URLs separately, I get a 403 error (permission denied: ACL denied) from lh4.googleusercontent.com, regardless of which IP I am using (i.e. I can reproduce this even when tunnelling through a VPN).
The pictures should be back up. They just got mis-linked while some restyling was going on. Thanks for the heads up.
Correct mapping of transcripts is a clear issue when converting variant descriptions using chromosomal/genomic coordinates to transcript coordinates. We have developed Mutalyzer’s Position Converter just for this purpose. We were using UCSC RefSeq mappings for its mapping database, because they were easy to get until one of our users pointed out that small exons were mismapped. We mentioned this BLAT problem to the UCSC a few years ago, but decided to switch to the NCBI mapping data used by MapViewer. The seq_gene.md file we use is available in the directory ftp://ftp.ncbi.nlm.nih.gov/genomes/MapView/Homo_sapiens/sequence/current/initial_release/
We still want to include mappings to ENSTs as well, but found out that versioning is an issue that will have to be solved first. Variant descriptions using ENST reference sequences without version number are ambiguous. They may be incorrect when used in combination with the next Ensembl release.
I’ve come across Mutalyzer a couple times now and I’ll have to try to put it to use on my next project. I really like that you expose an api to the service!
We are actually in the process of curating NCBI’s GFF files directly to avoid some of the mapping problems as well. It’s looking promising, so be on the look out for the track to be available soon in GenomeBrowse.
Very useful post, Andrew. I’m seeking for a good annotation tool for my sequencing project. Actually I found a very latest one which called Combined Annotation Dependent Depletion (CADD). It’s a very nice tool which can integrates dozens of annotations into one metric by contrasting variants that survived natural selection with simulated mutations. Do you have any comments on this fancy new method?
Thanks,
Meijian
CADD is extremely interesting! The original paper is well worth reading, if you haven’t had a chance to take a look at it yet — http://www.nature.com/ng/journal/v46/n3/full/ng.2892.html.
CADD is trying to deduce the how damaging a particular variant is based on numerous annotations, including VEP. As such it’s really a second level of analysis. I think machine learning methods, such as the support vector machine CADD implements will become more and more prevalent in the next few years, especially as variant datasets grow to a sufficient size to be useful for training these algorithms. It is definitely an area of bioinformatics that I think is ripe for innovative research.
Thanks for your article Andrew, very useful indeed.
Do you have any comments on performance and memory usage? It seems to me that both annovar and SnpEff are not top performers at these points, didn’t have the opportunity to check vep.
Thanks!
Pablo.
Hey Pablo-
I didn’t really run them in comparable performance settings. I ran VEP and ANNOVAR via their web interfaces and SnpEff on my MacBook. It’d be interesting to benchmark for sure!
Great post Andrew – I’m just stepping in the world of variant annotators and your post is a valuable reading! We have VEP and SNPeff activists in our team, but now have to chose one or the other for our generic pipeline. I’m also curious about the answer to Pablo’s question regarding performance. If both VEP and SNPeff perform equally well for our purpose, then performance could be the discriminating factor.
Dear Andrew,
Thanks for the nice post!
I am the author of ANNOVAR, and a number of users asked my opinion on this blog post over the past a few months. I finally decided to post a comment now only because (1) I got complaints that Golden Helix sales persons use this blog to attack on Annovar/VEP/snpEff inappropriately (2) early November 2014, your Director of Marketing began to send mass emails to researchers solely to advertise this particular blog post, potentially presenting misleading information to a very large number of scientists.
There are 10 places where Annovar was mentioned, but the information may not be accurate enough. To straighten the records, I respond to them one by one below:
1. “Annovar’s output is a tab separated file, while SnpEff and VEP produce VCF files which use the “INFO” field to encode their annotations. ……. Annovar’s rows changes depending on context. For example, Annovar uses the gene field to provide distance information for all intergenic variants. Similarly, for variants labeled as splicing, it overloads the gene field with HGVS notion…… I’d argue that the ability to easily parse the output file should be paramount when designing the tool.”
This statement is not accurate, and may be due to the lack of familiarity with Annovar: (1) Annovar (table_annovar) can certainly use INFO field to encode annotations in VCF file, exactly like SnpEff and VEP. (2) Annovar (table_annovar) can also generate tab-delimited output that is easily parsable without any of the aforementioned problems, and in fact it is far more parsable than VCF (3) the annotate_variation generates gene-based annotation output that is less parsable (as mentioned); however, Annovar was developed in a time when MAQ and Complete Genomics and SOAPsnp and SOLID BioScope and Illumina Casava each generates different formats for variant calls (and none of them uses VCF). For the purpose of backward compatibility for old users, I tried not to change output format, yet adding such parsable functionality in table_annovar instead. It is merely a design decision to avoid drastic changes to something stable.
2. “Moreover, some genes overlap so a single variant could affect multiple transcripts on multiple genes. Both SnpEff and VEP list each way the variant was classified. Annovar instead returns only the most deleterious variant based upon a priority system.”
I am not sure exactly what you mean, but regardless, it is not accurate: (1) if multiple transcripts/genes cover a single variant, they will all be reported in Annovar (2) adding -separate will be able to generate all predictions, not just the most deleterious predictions.
3. “Another interesting decision Annovar made is not to annotate variants that lead to start loss/gain mutations. While I can see the reasoning behind calling a start gain just a nonsynonymous variant, not annotating start losses is troubling.”
Annovar does annotate start loss; however, instead of calling it “start loss”, it calls it as whole-gene given that the entire transcript cannot be translated. It makes sense either way.
4.” SnpEff, uses 5kb to define upstream and downstream regions, while Annovar uses 1 kb. In many other cases, variants in non-coding regions were bucketed into the “ignored” category.”
This can be adjusted in Annovar via an argument –neargene in command line.
5. ” 7:117120154-117120154 -/T” (frameshift vs stopgain)
Most frameshift mutations within exons will lead to premature stop codon (stop gain). In Annovar, one can use coding_change.pl to infer the new protein sequence to see where the new stop codon is. Here the mutation is referred to as “frameshift” and there is nothing wrong about it. In general, “stopgain/stoploss” in Annovar only refers to the situation where a SNP results in gaining/losing a stop codon at that specific codon only.
6. ” 7: 117105836-117105838 TTT/-” (upstream vs NC exonic)
It helps if you can check this region is UCSC Genome Browser. There is no gene in this region from RefSeq gene and UCSC gene, but there is a “ensembl gene prediction” here that this is a non-coding transcript for CFTR. CFTR is absolutely a protein coding gene. The logic for Annovar is described in detail here: http://www.openbioinformatics.org/annovar/annovar_gene.html. If a protein-coding gene was annotated with a non-coding transcript then this transcript will be ignored. Thus this mutation is treated as a intergenic mutation. It just makes sense doing it this way.
7. 7: 117307160-117307161 T/- (frameshift vs stoploss)
Annovar calls it stoploss rather than frameshift only because the stop codon at that specific location is lost, as I mentioned above.
8. 7:117119513 -/A (splice site variant)
If you read Annovar documentation, by default only bases within 2bp of intron/exon boundary is referred to as splice variants (but -slicing argument can be used to change this behavior). As this position is 3bp away from boundary, it is called as intron by Annovar. VEP uses 5bp I think, so it is called as splice_site_region. Nothing surprising here.
9. 7:117144417 -/ATG (splice site vs stopgain)
It is a splice site, but since a stop codon is inserted right at that site, it is called as stopgain by Annovar, as I mentioned above.
10. 7: 117307124 – 117307126 GAG/- (frameshift vs inframe)
If you read the figure very carefully, you will see that this “first transcript” is clearly an annotation error. A codon, by definition, must have three nucleotides, whether or not this is a stop codon. So Annovar ignores this wrong transcript annotation, and uses the other correct transcripts instead, and correctly calls this mutation as nonframeshift deletion. It just makes sense.
In summary, I do not see anything wrong with Annovar’s results, based on all the examples that you have presented above. In addition, I clarified a few imprecise statements about Annovar, to avoid misleading readers of this blog. Nevertheless, I thank your efforts doing deep research on this area; after all, we know that all software has bugs and it is the comparison against each other that helps improve tools. Spending efforts and time doing extensive benchmarking certainly should be applauded greatly.
Best regards,
-Kai
Hey Kai-
Thanks so much for taking the time to read the post and comment on ANNOVAR specifically. I in no way wished to attack any tool and was more interested in a comparison to see if I could infer what specific genomic features give annotation software problems. Since I had just started to write our own variant annotator, I was especially curious to figure out what difficulties I should expect. Having just completed the first version, I can say that implementing these types of algorithms is no easy task! I’ve never dealt with so many (literal) edge cases (utr boundaries, tx boundaries, and splice sites, to name a few). As I said toward the end of the post, it is extremely important for the user to become familiar with both the annotation software and transcript set being used. I’d go so far as to say that this experience is probably more important than the choice of tool!
I should have been more clear that I was using the (now deprecated) wANNOVAR2 site for my ANNOVAR runs and the output that I received was the annotate_variation as mentioned. It sounds as if table_annovar is just what I was looking for! As I mentioned, parsing the VCF’s generated by VEP and Snpeff wasn’t easy either–I just had a better strategy for parsing the INFO field. And the annotate_variation output is what gave me the impression that all ontologies were condensed per gene.
Annotating start losses the way you describe makes a lot of sense. I would still say using the term start_loss has some merit if only to minimize differences in semantics.
Finally, in terms of the the individual variant comparisons, your comments highlight the difficultly in choosing which otological term to use to label a variant. I think this is a point that is often taken for granted by end-users of these tools. In many cases, it may not make much difference during analysis since a researcher would probably group both frameshift and stop-loss variants into the same impact bucket (LoF). However, for other ontologies, as you rightly point out, it does pay to be aware of if splice variants are defined as 2 bases intronic or 8 bases intronic, since the functional impact could differ greatly.
I think ANNOVAR makes a lot of really smart choices, many that you highlight above. I also want to point future readers to the new hosted version of ANNOVAR at http://wannovar.usc.edu/. It should be noted that the Phenolyzer tool offered in conjunction is something that neither VEP or SnpEff offer. I’m excited to run some data through it!
Thanks again for you insights!
Hi Andrew,
Thanks for the useful post with lots of information. I have a comment regarding your example of Splice Site vs Stopgain @ 7:117144417 -/ATG and the statement
“The first two intronic bases (GT) act as signal for the intron to be spliced out. However, the stop codon is inserted just before these two bases. Thus, during translation the ribosome will hit the stop codon and stop producing the protein rather than splicing the two exons together.”
Exon splicing happens before translation, and does not involve ribosomes. These are two different processes. Ribosomes will translate from a final form of mRNA, with introns already spliced out (this might be relevant to your example about TNNI3, although I did not think much about it). Also, I think the stop codon is NOT inserted just before GT, but it will presumably be TGA, where A will presumably be the first base of the next exon (not seen on the picture). ATG is not a stop, but a start codon, and it would be out of frame where it is inserted.
Thank you.
Hello,
I have a question concerning variant annotation results (With program VariantAnnotation)
How a variation (SNPs) can be an intron and exon at same time? It is by frameshift?
Example:
seqnames position width strand LOCATION QUERYID TXID CDSID GENEID
scaffold_8 1461234 1 – coding 830 40443 154081 Eucgr.H00154
scaffold_8 1461234 1 – promoter 830 40443 NA Eucgr.H00154
Thank you for your very comprehensive overview. What would you personally use for a clinical setting (identifying disease causing variants in cohorts)? SNPeff + Ensembl + excl. of incomplete transcripts? Best, Chris
Hey Chris,
Since we wrote this, we built our own variant annotation algorithm in VarSeq that we think combines the best of SNPeff and Ensembl. It also handles incomplete transcripts, picks out the clinically relevant ones and is focused on providing all the appropriate other annotations necessary for the clinical setting (many which are not available in these tools).
Check out VarSeq and feel free to let us know if you want to take a closer look with your own data by requesting an evaluation.
Hi Gabe,
I’m new to the annotation side of things and come more from a clinical background, so forgive me if my question(s) see a bit basic. Trying to learn and figure out which annotation software(s) to use and why, especially n a clinical setting. We’ll be sure to check out VarSeq. I understand that HGVS provides for the 3’rule and SNPeff adheres to these specs. Are there any advantages to SNPeff over other annotation tools? In cases where the reference-transcript alignment contains indels are there any advantages or differentiators from other tools?
Thanks for any input.
Best,
Todd
Hey Todd,
Yes, we adhere to the 3′ rule in VarSeq as well.
The reference-transcript alignment issues are not auto-corrected by any of the bulk variant annotation tools that I know of. We are actively looking at getting the types of data about the reference-transcript alignments (such as what were the missing bases from the reference) that could allow us to do this type of auto-correcting.
Some web-based tools like the NCBI variant annotator can handle this.
Thanks,
Gabe
Thanks for the great article. It inspired and guided our thinking on this subject as we worked on it as well.
Our group has just published a preprint at biorxiv on the subject of variant annotator comparisons. We provide a python framework for doing automated comparisons as new methods become available:
http://www.biorxiv.org/content/early/2016/09/30/078386
I don’t usually comment but I gotta admit regards for the post on this amazing one :D.
Thank you for the excellent post! This is a very nice piece of investigation.
Would you mind sharing the simulated CFTR VCF files annotated with the three tools you mentioned?
Hey Alex,
We put all the analysis and data wrangling code for this blog post on our github here: https://github.com/goldenhelix/cftr-variant-classification-analysis
Thanks, Gabe
Great, thank you very much. We’ll put it to good use.
We received the results for sequences of exon 7 from APOBEC3G gen, and we found a change (rs369306100) that be present in all sequences (240 samples), however, this change (c.1109A>C), has a MAF very low: C=0.00005/6 (ExAC) C=0.00008/1 (GO-ESP) C=0.00008/10 (TOPMED) and others. We found this change in 240 samples from Colombian people (n=240). The sequence is clear, we don´t have unpecifics products in PCR, and the electropherogram is very clear and clean. The products were sequencing in two different opportunities and we had the same results. It is possible that you can help us with this unexpected result? Can exist an error in this annotation? Can you give us some indication to understand these results? We appreciate your help. Thanks for your time. We will be waiting for your answer.
Thanks a lot.