VCF file format comes with a lot of interesting quality assurance and statistics fields that can be used for filtering in VarSeq. Open your files in a text editor to see all the fields that are available in your files, each field will have a header line with a description of its content. See the VCF Specifications to help with the interpretation of the information.
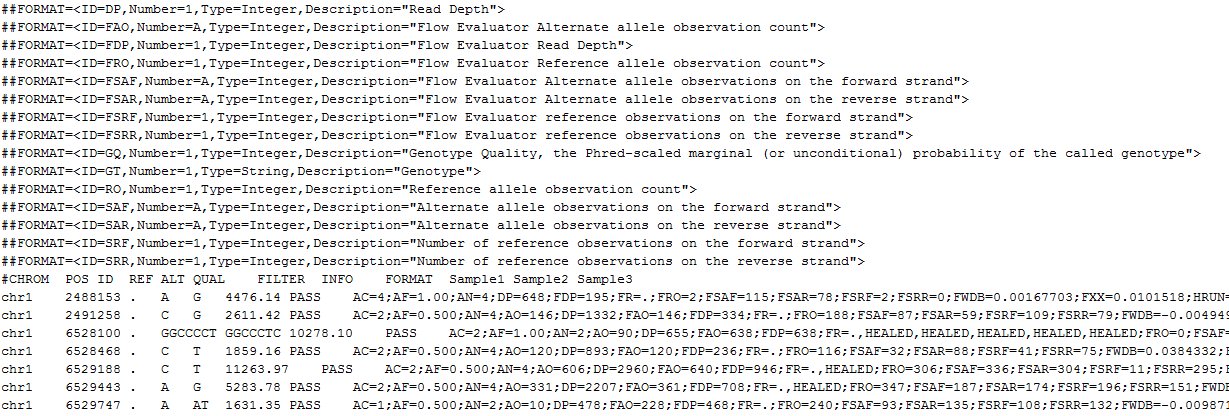
One of the most used values for filtering variants in a somatic mutation workflow is the Alternate Allele Frequency for each sample. This field is not always provided directly in the VCF data but don’t worry, VarSeq will automatically calculate the frequency using the provided allelic depth fields in the file.
Depending on the Variant Caller that was used to produce your files the allelic depth information can come from a variety of fields within the VCF file and VarSeq can use them to compute the Alternate Allele Frequency (Alt Allele Freq).
We will first look for observed counts for both the reference and alternate alleles, these values will be provided in the AO and RO fields. They can also be available as Flow Evaluator observed counts in the FAO and FRO fields (Flow Evaluator fields are preferred).
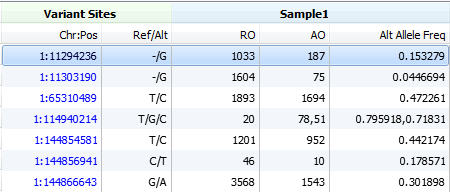
Next, we will look for observed alternate allele counts and the total allelic depth fields, the alternate allele counts will once again come in either AO or FAO fields. The total allelic depths will be found in the DP or FDP fields respectively.
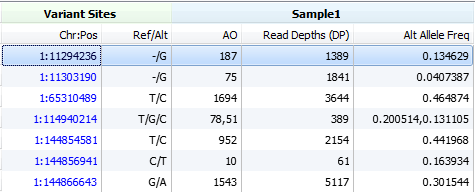
If none of the above fields are available we will then use the unfiltered counts for all reads that carried reference or alternate alleles found in the AD field. This field is an array where the first entry represents the reference allele and then the following entries are for each alternate allele at this locus.
As a last resort, VarSeq will look for the DP4 field which can commonly be found in VCF files prepared by SAMTools. This field has four entries in the following order: forward reference count, reverse reference count, forward alternate count and reverse alternate count.
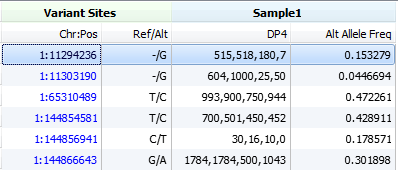
If your data contains this information in a different field or format then you can compute your own alternate allele frequency using the Add > Computed Data… > Compute Fields algorithm. If you have questions about the computation or need assistance computing your own field just send us an email at [email protected]!




Hello Jami,
Its a very informative post with exact cases and illustrations. In first snapshot header line DP denotes description as “Read depth” in the vcf file.
I have 2 vcf files to calculate alt allelle frequency as :
A) In vcf file generated using samtools, DP description is reported as “Raw Depth”. In this case DP4 field exists in the vcf file.
B) In vcf file generated from GATK DP description is reported as “Filtered Depth”. In this case AD field exists in vcf file.
Would there be different formula to calculate altered allele frequency where DP field has such different meanings.
Hi Vaibhav,
I am glad you found this post helpful!
The DP field will only be used in the calculation if there is an associated AO (observed alternate count) field and no consideration is made for whether the DP field is contains filtered depth or raw depth counts. In the case of your SAMTools VCF file if there is no AO field available then the DP4 field will be used, similarly for your GATK VCF the AD fields would be used for the calculation.
Let us know if you have any further questions.
Thanks,
Jami…
Hi Jami,
Thanks for the post. I came across it while searching for an explanation of alternate allele. I am new to the field and maybe for that reason could not fully understand the concept of alternate allele. Is is the allele found in the reference genome in case it is different in the sample? For example, if the sample had the allele A, and if the reference genome has a G, then is G an alternate allele? I would greatly appreciate if you could clarify with an example.
Thanks,
– Pankaj
Hi Pankaj,
I am happy to help with your questions.
The alternate allele is any allele not present in the reference sequence at a particular location in the genome. So for your example if the allele for your data was an A and the reference genome has a different allele G at the reported location, then your data has the alternate allele (A) as the reference (G) is different. The alternate allele is sometimes referred to as the “variant” allele or the “mutated” allele, as it is the allele that varies from what is reported in the reference genome.
Let me know if you have any further questions.
Thanks,
Jami…