We are ending the first half of the year with a webcast featuring our entire clinical software stack: Golden Helix’s End-to-End Solution for Clinical Labs. This webcast is scheduled for Wednesday, June 7th at noon EST, and it’s not one you’ll want to miss! Here are the full details: Wednesday, June 7th 12:00 pm EST In this webcast we will provide an… Read more »
Yesterday we had an excellent webcast presentation featuring Sentieon’s Dr. Donald Freed. Golden Helix recently partnered with Sentieon to integrate their secondary analysis tools into our software stack, and this presentation is an introduction to their genomic tools. Please check the webcast out here if you missed it! There were a number of questions asked in the live event, so in… Read more »
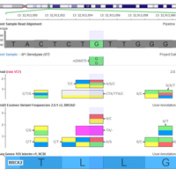
Annotating with gnomAD: Frequencies from 123,136 Exomes and 15,496 Genomes When the Broad Institute team lead by Dan MacArthur announced at ASHG 2016 that the successor to the popular ExAC project (frequencies of 61,486 exomes) was live at http://gnomad.broadinstitute.org/, I thought their servers would have a melt-down as everyone immediately jumped on and started looking up their favorite genes and… Read more »
The new VSReports tutorial covers a basic VSReports workflow with an emphasis on understanding and exploring report customizations. This tutorial requires an active VarSeq license with the the VSReport feature included. You can go to Discover VarSeq or email [email protected] to request an evaluation license with the VSReports functionality included. VS Reports provides the ability to generate clinical-grade reports. This feature enables VarSeq… Read more »
We are excited to announce our next webcast, The Sentieon Genomic Tools – Improved Best Practices Pipelines for Analysis of Germline and Tumor-Normal Samples. This webcast is scheduled for Wednesday, May 17th at noon EST and will focus on our new partnership with Sentieon. Wednesday, May 17th 12:00 pm EST With next-generation sequence datasets frequently reaching petabytes in size, processing genomic data accurately… Read more »
Today, we are happy to announce a multi-year partnership with Sentieon, a company that develops bioinformatics secondary analysis tools to process genomic data. This partnership will integrate Sentieon’s secondary analysis tools with Golden Helix software to provide users with a comprehensive solution for genomic data analysis. Sentieon’s suite of secondary analysis tools made the significant improvement in runtime over BWA-MEM, GATK,… Read more »
The new VSWarehouse Tutorial covers the basic VSWarehouse workflow.This tutorial focuses on connecting to a VSWarehouse instance from VarSeq, adding an existing VSWarehouse project as an annotation source and using reports and assessment catalogs hosted on VSWarehouse. This workflow requires an active VarSeq license with the VSWarehouse feature included. You can go to Discover VarSeq or email [email protected] to request an… Read more »
We had lots of customers publish their work using our SVS software, and I wanted to share their work with you. Congrats to all! Here are some of the highlights: Francesca Fernandez of the University of Wollongong along with colleagues published Effects of common GRM5 genetic variants on cognition, hippocampal volume and mGluR5 protein levels in schizophrenia in Brain Imaging and… Read more »
Nathan Fortier’s webcast yesterday, CNV Analysis in VarSeq – A User’s Perspective, was an excellent demonstration of our CNV capabilites, so check it out if you missed it. There were a number of questions asked in the live event, so in today’s blog post I wanted to recap the Q&A session. Question: Do the reference samples have to be from the same… Read more »
Earlier this year we released our own optimized and integrated BEAGLE implementation for SVS based on the BEAGLE 4.1 and optionally 4.0 algorithms. One of the commonly requested features since that released was to expand the algorithm implementation to be considerate of the parent-offspring relationship between samples to inform and improve the accuracy of the haplotype phasing. With this information,… Read more »
In our latest VarSeq release, we updated our PhoRank algorithm with the ability to specify OMIM phenotype terms not present in HPO, as well as a general update to the algorithm to improve the results. In this post, we review the fundamentals of how PhoRank determines the ranking of genes in your VarSeq projects based on your input phenotype terms… Read more »
Recently, we added a natively supported Genotype Phasing and Imputation capability in SNP & Variation Suite 8.7.0. Since then we have had fantastic feedback and adoption as folks take advantage of the BEAGLE 4.0 and 4.1 algorithms from within their existing SNP GWAS and agrigenomic workflows. One piece of feedback we heard from our time at PAG, ACMG and our… Read more »
We are pleased to announce our next webcast, CNV Analysis in VarSeq – A User’s Perspective. The live event is is scheduled for Wednesday, April 19th at noon EST. Here are the specifics: Wednesday, April 19th 12:00 pm EST Clinical labs must have the ability to go from a collection of samples to a professional report documenting a short list… Read more »
It may be possible to say that annotating a variant correctly and accurately against gene transcripts is the most important job of a variant annotation and interpretation tool. We take it very seriously at Golden Helix as we support VarSeq and its use by our customers in both research and clinical contexts. It has been a source of frustration that… Read more »
Last week was fast and furious for those of us who made it to Phoenix attending ACMG 2017. The event was exceptionally well attended, with great talks and sessions. Here are some of the highlights that are worth mentioning from our perspective. Numerous talks referenced how important the updated Exac/gnomAD database is for clinical NGS analysis. The new dataset includes… Read more »
Join our upcoming webcast : Wednesday, April 5th 12:00 pm EST Dr. Reza Sailani is a Research Fellow in the Genetics department at Stanford University. To provide an overview of his research, Sailani will present on the following two recent studies he has conducted: Association of AHSG with alopecia and mental retardation (APMR) syndrome: Alopecia with mental retardation syndrome (APMR) is… Read more »
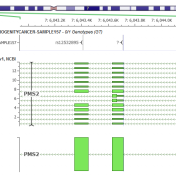
Today, I am happy to announce the introduction of VS-CNV for Gene Panels and Exomes. We have developed this capability in partnership with PreventionGenetics. PreventionGenetics will use VarSeq CNV for analysis of gene panels, and in the future for exome sequencing. The software gives PreventionGenetics the opportunity to conduct a comprehensive CNV analysis on NGS data, in many cases eliminating the need… Read more »
ACMG 2017 is just around the corner! We are halfway through March already and it’s just about time to head off to sunny and warm Phoenix, Arizona. While the temps have been mostly mild for the last few weeks in Montana, I bet those of you in the northeast are looking forward to your time in Phoenix! You will find… Read more »
Ever since the MacArther Lab announced the new gnomAD browser at last year’s ASHG conference, we have had many requests from our customers to make this new variant frequency source available within both VarSeq and SVS. This new dataset includes variants obtained from 123,136 exome sequences and 15,496 whole-genome sequences. In comparison to the original ExAC dataset which contained exomes… Read more »
In the past couple of weeks, the topic of the Filter and Quality fields in the popular ExAC population catalog has come up a number of times. It turns out that unlike the 1000 Genomes project, which decided to very heavily filter their variant list to only contain variants they consider high quality, ExAC chose to include more dubious variants… Read more »