Many thanks to those who came to view our most recent webcast, From Panels to Genomes with VarSeq: The Complete Tertiary Platform for Short and Long-Read NGS Data. This was a great opportunity for us to showcase the breadth of our workflows, from short-read to long-read, panels to genomes, singleton analysis to families. In this blog, we would like to briefly recap our feature highlights, along with answering some of the questions our viewers left during the webcast.
Cancer Panel Workflow
Our first use case highlighted a single sample TSO500 workflow. With this workflow, we were able to leverage different cancer databases such as CIViC, Golden Helix CancerKB, and more to isolate cancer-specific variants. By using a virtual gene panel, we were able to narrow the scope to variants specific to our cancer type, non-small cell lung cancer. Last, in our filter chain, we brought in our brand new Cancer Classifier to look for Oncogenic and Likely Oncogenic variants of interest.
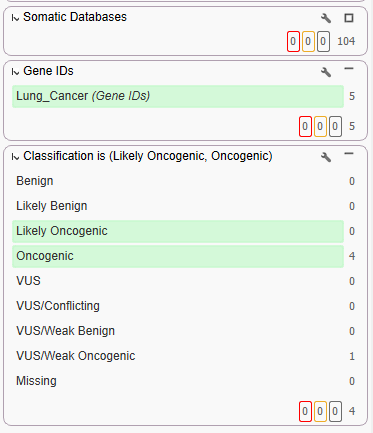
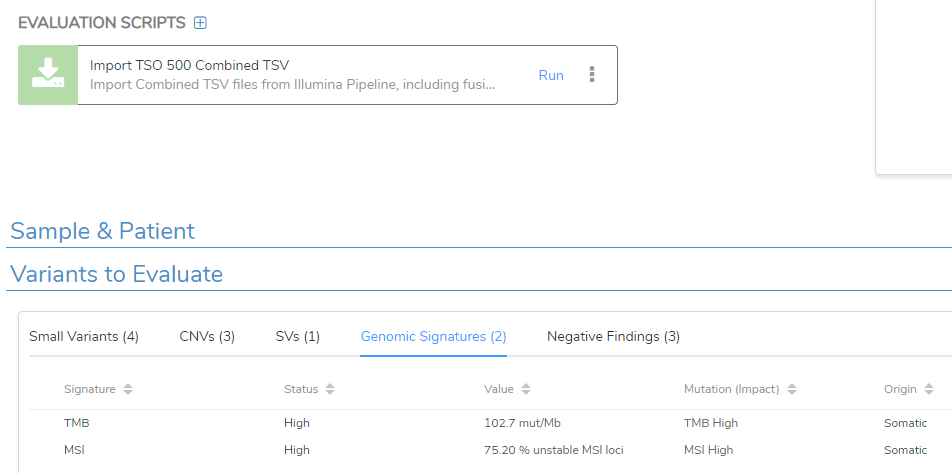
When working with cancer kits like TSO500, Archer, or others, we may have externally sourced data we want to add to our analysis pipeline. We have used the evaluation script to bring in the combined TSO500 Genomic Signatures, CNVs, and Structural Variants, as seen below (Figure 2). All of these variants and genomic signatures contributed to the overall drug recommendations we rendered in the final clinical report.
Germline Whole Exome Workflow
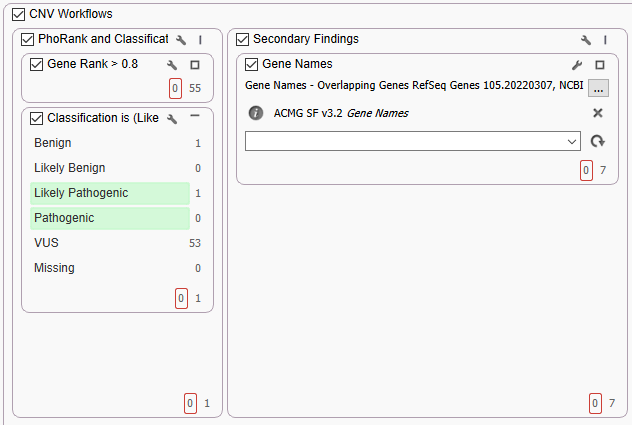
The next workflow from the webcast was the single sample germline whole-exome workflow. This project, unlike the project above, used the VarSeq CNV caller to generate the CNVs. Here we can show parallel workflows for not only the variant filter chains but also our CNV filter chains (Figure 3). This parallel filter chain leverages a specific phenotype to narrow the scope of CNVs or variants to those relevant to the patient. This is done with our PhoRank algorithm. Next to the phenotypic prioritization, we are searching for any secondary findings by using the ACMG Secondary Findings gene list as a virtual gene panel.
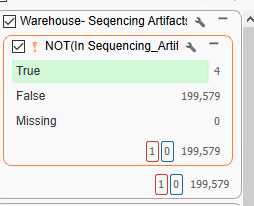
The other major takeaway from our single sample workflow is the fantastic utility of VSWarehouse. VSWarehouse is the ultimate solution for saving data from your VarSeq Projects. With this tool, we are able to upload variant frequencies from your samples, create overarching variant and CNV catalogs, annotate your projects from your VSWarehouse database, search for changing classifications, and more. Here, we have an example where known sequencing artifacts have been recorded in VSWarehouse (Figure 4). By annotating the current project with this catalog, we can build the artifacts into the filter chain to automatically scrub out any known artifacts from consideration. This utility can be extended to CNV calls as well. Another example shown was how our singleton workflow was annotated with the contents of a variant truth set project. We could leverage the frequencies of the truth set to search for variants unique to our single whole exome sample.
PacBio Long-Read Trio Workflow
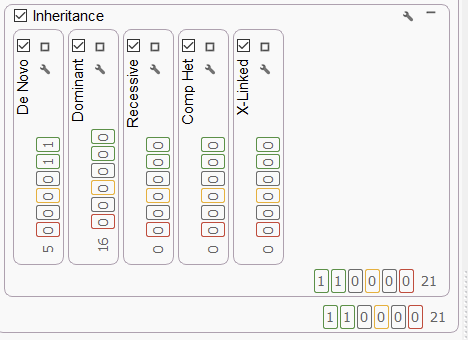
We were most excited to share our PacBio HiFi long-read project with you on this webcast. This project started with a staggering 7,096,329 variants across our mother, father, and proband samples, and our comprehensive filtering strategy reduced the remaining variants to 21 without a phenotype or just one variant with a phenotype application. As seen below (Figure 5), our inheritance filters are able to parse out the remaining variants into bins for De Novo, Dominant, Recessive, Compound Heterozygous, and X-Linked for easy analysis.
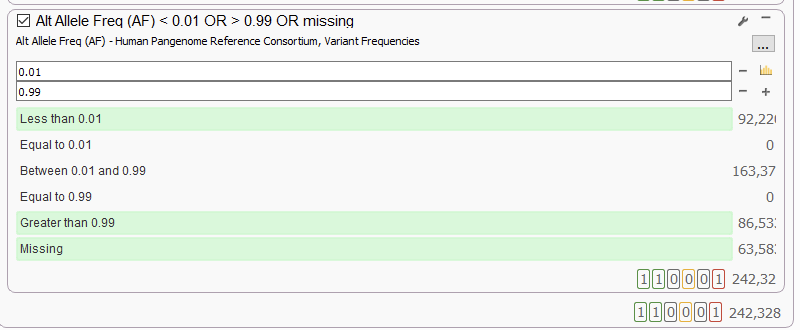
We are also thrilled to announce the recent PacBio Human Pangenome Reference Consortium (HPRC) release for variant filtering (Figure 6). In our experience, this annotation source, when applied to PacBio HiFi sequencing, does a great job cutting through the ‘noise’ of common variant calls. In addition to the HPRC for variants, we now host the HPRC annotations for CNV frequencies and fusions as well.
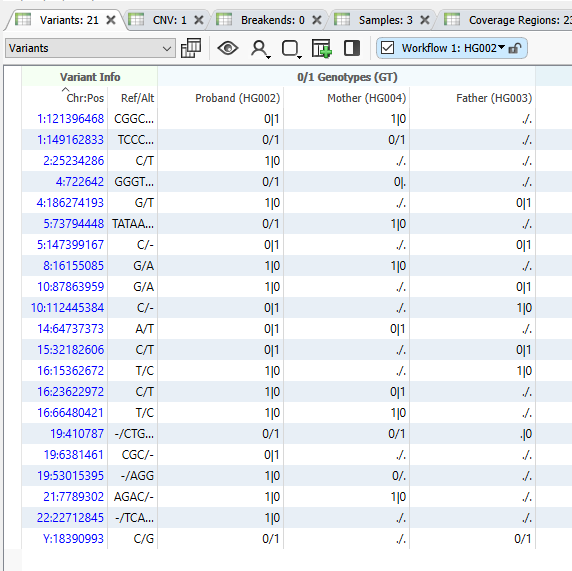
Our last major takeaway with our PacBio HiFi Long-read Trio is the addition of the phased variant calls. As seen below (Figure 7), adding the | to the GT field now helps us understand the variant’s position on a given chromosome. This is a huge boon to compound heterozygous detection.
Questions
Our webcast viewers had several lingering questions we wanted to address here.
1. PhoRank: Can we input multiple phenotypic terms into a search, or just one at a time?
Yes! You are able to add many phenotypic terms to your PhoRank search. When there are multiple terms, they can act together to contribute to a PhoRank score, and this can help you find the best overall matches.
2. Can you only take PacBio VCFs, or can you take other long-read VCFs?
As long as the data is in a standard VCF format, VarSeq does not care about the Secondary Caller of origin. We can look at short-read data from other callers like Sentieon, GATK, and more. We are also able to look at long-read VCFs from other companies as well.
3. Can VarSeq be used to analyze a cohort, or is this only for clinical applications?
We do support Cohort Analysis, and that is a growing use case among our customer base. We have a number of algorithms to help detect variants unique to affected individuals, to look at variant frequencies across a cohort, a specific version of PhoRank for cohorts, and more!
Thank you all for attending our webcast. If you have any lingering questions, please reach out to our FAS team at [email protected], and we will be happy to help you with your NGS analysis needs.



