About Gabe Rudy
Meet Gabe Rudy, GHI’s Vice President of Product and Engineering and team member since 2002. Gabe thrives in the dynamic and fast-changing field of bioinformatics and genetic analysis. Leading a killer team of Computer Scientists and Statisticians in building powerful products and providing world-class support, Gabe puts his passion into enabling Golden Helix’s customers to accelerate their research. When not reading or blogging, Gabe enjoys the outdoor Montana lifestyle. But most importantly, Gabe truly loves spending time with his sons, daughter, and wife. Follow Gabe on Twitter @gabeinformatics.
Since we released our Phenotype Gene Ranking algorithm in VarSeq, it has become a staple of the way people conduct their analysis. It allows for a combination of filtering with ranking to prioritize follow-up interpretations of analysis results. Our PhoRank algorithm will be available in our upcoming SVS release to also aid in the numerous research workflows performed on SNPs… Read more »
It may have been easy to miss in the drum-beat of monthly annotation updates we do here at Golden Helix, but there are a couple of things that are very special about the January update to the ClinVar database: We added new fields including HGVS names of variants and citations in PubMed for variants ClinVar nearly doubled in size by… Read more »
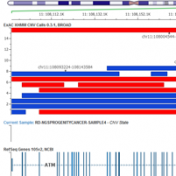
ExAC CNVs were released publicly with a recent publication, providing the full set of rare CNVs called on ~60K human exomes. While there are many public CNV databases out there, this is the first one that was derived from exome data, and thus includes both extremely rare and very small CNV events. With the recent release of Golden Helix’s CNV calling… Read more »
While clinical assessments of germline mutations have been collected in ClinVar under the stewardship of the NCBI and the collaborate effort of many testing labs, the same type of resource has been missing for mutations that could informal clinical care in Cancer. Or at least, that is what I thought until I started to work with CIViC. With the stewardship of… Read more »
One of the tools at the top of the toolbox for researchers working with microarray data is genotype imputation. Genotype imputation is the process of inferring the genotype of one or more markers based on the correlation pattern (aka linkage disequilibrium or LD) of the surrounding markers for which genotypes are known. We have now integrated a natively ported version of BEAGLE into Golden… Read more »
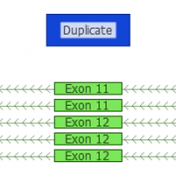
Copy Number Variants have been important to clinical genetics for quite a while now. So, what has made now the right time to be looking at calling CNVs from NGS data? Well, there are a number of good reasons. The dominant one is simply that the NGS data you are already creating for calling variants can be used in many cases… Read more »
Big data is here, but fear not, you don’t need a Hadoop cluster to analyze your genomes or your cohorts of tens of thousands of samples! It turns out, for the kind of algorithms employed in variant annotation and filtering, running optimized local programs is often faster anyway. As we support our diverse customer base, we have definitely seen the… Read more »
Now available in SVS! Increasingly important in the analysis of the genotype to phenotype relationship is accurately accounting for the relatedness of samples. This is especially important to model correctly in plant and animal populations where man-directed breeding shapes the relationship structure. Along with trait association, one of the high-value use cases for genotyping animals and plants is to estimate… Read more »
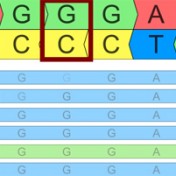
Variant Normalization: Underappreciated Critical Infrastructure It may surprise you to learn that every variant in the human genome has an infinite number of representations! Of course, although true, I’m being a bit hyperbolic to prove a point. Even seemingly simple mutations like single letter substitutions are legitimately represented differently in the local context of other mutations that can be described… Read more »
I’m very glad I had the chance to attend ESHG 2016 in Barcelona and talk to so many people about Golden Helix and our software at our booth. ESHG may be the little sibling in size compared to ASHG, but my impression is that it punches above its weight in terms of advancing human genetics applicability to human health and… Read more »
There used to be much energy expended at conferences, bioinformatics forums and even publications about what was the better strategy for interpreting variants of clinical significance: Rule-based filtering and classification mechanisms or rank-based prioritization through all-encompassing “pathogenicity” scores. Both have shown to be effective. Rule-based systems, as exemplified in this filtering diagram in Baylor’s ground-breaking paper on clinical whole-exome sequencing… Read more »
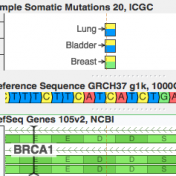
ICGC’s database is now available For quite a while, COSMIC has been synonymous with the catalog of “known somatic mutations”. It is the ClinVar of cancer mutations and invests heavily in “expert curation” (having human experts read papers and pull out references to published somatic mutations). But it turns out there is a new kid on the block, and he… Read more »
Scaling is in our DNA: Making Genomics Accessible One of the things I absolutely love about the work we do at Golden Helix is keeping up with the changes in data analysis driven by the iterative and generational leaps in technology. But one thing has always been a constant since day one: we break preconceived notions of what scale of… Read more »
Yesterday, it was my pleasure to share in a live webcast our integrated solution for genetic data warehousing, VSWarehouse. If you missed the webcast live, feel free to check out the recording. Although we had a great set of questions at the end of our presentation, we didn’t have time to answer all of them, so here is a selection of… Read more »
There is no doubt that we have big data in the field of genomics in general and Next Generation Sequencing specifically. Illumina’s latest HiSeq X can produce 16 genomes per run, resulting in terabytes of raw data to crunch through. Yet all that crunching is not the hard part. So, what is the main obstacle to scientists being able to… Read more »
Probably one our most popular public annotation sources we curate and update is the database of Non-Synonymous Functional Predictions (dbNSFP). In it’s recent update, it has expanded the predictions to include FATHMM-MKL and VarSeq now incorporates this new prediction into its voting algorithm of now 6 different discrete predictions per variant. You can update to dbNSFP 3.0 using the built-in… Read more »
As VarSeq’s adoption has grown among analysts using whole exome data to diagnose rare diseases, a couple of family designs outside of the common trio of an affected child and both parents have come up frequently. While having both parents provides the maximum power to discover de novo mutations and recessively inherited variants, it is not always possible to contact… Read more »
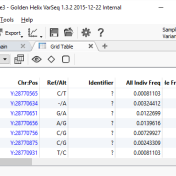
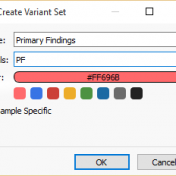
With the release of VSReports, we added the ability to “select” rows of your filtered output (often variants, but potentially things like coverage regions or genes) with a new feature dubbed “Record Sets”, but more often described as “colored checkboxes” for your tables. Although necessary for the important task of marking primary, secondary or other sets of variants for a… Read more »
When it comes to down to it, the genomic variants we collect in a research and clinical setting are impossible to interpret without that important link of how genes are related to phenotypes. Indisputably, the Johns Hopkins project to catalog all evidence related to inheritable Mendelian diseases is our best repository of this evidence. Online Mendelian Inheritance in Man (OMIM)… Read more »
Have you ever scratched your head when looking up a variant and it seems like the number you have for its position is one off from what it looks like in the file or database? You may be running into the dreaded world of 1-based versus 0-based coordinate representation! If it’s any consolation, I can promise that all the bioinformaticians… Read more »