About Gabe Rudy
Meet Gabe Rudy, GHI’s Vice President of Product and Engineering and team member since 2002. Gabe thrives in the dynamic and fast-changing field of bioinformatics and genetic analysis. Leading a killer team of Computer Scientists and Statisticians in building powerful products and providing world-class support, Gabe puts his passion into enabling Golden Helix’s customers to accelerate their research. When not reading or blogging, Gabe enjoys the outdoor Montana lifestyle. But most importantly, Gabe truly loves spending time with his sons, daughter, and wife. Follow Gabe on Twitter @gabeinformatics.
This webcast generated a lot of great questions about the content covered in the video above. I have summarized our Q&A session below and included some questions I didn’t have time to answer during the live event. If you have any further questions, reach out to us at info@goldenhelix.com! Q: Can I upload my existing classifications into a consortium source?… Read more »
We just got back from three busy days at the Molecular Pathology (AMP) conference in friendly San Antonio, Texas. Keeping up the Golden Helix conference momentum for the year, we had 3-4 in-booth demonstrations a day covering our CNV calling, variant interpretation, and data warehousing products for NGS-based genetic tests. And in short, NGS based tests for cancer and germline… Read more »
VarSeq Stable 2.1.0 is Ready for Clinical Validation, See it in Action Next Week This week we are happy to see the general availability of VarSeq 2.1, the culmination of the last five months of work since we launched VSClinical. We have been blown away by the adoption of VSClinical, outpacing any previous product launch in terms of the pace… Read more »
This webcast generated some great questions! If you have any other questions for me that are not answered below, please feel free to ask those by emailing support@goldenhelix.com. Does VSClinical come with support for the new reference genome? Yes! We worked hard to make everything work in VSClinical regardless of your choice of reference genomes. The only caveat is that… Read more »
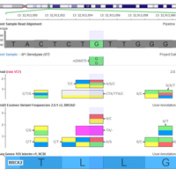
Streamlining the ACMG Guidelines and Providing Scoring Recommendations As we discussed in our recent webcast on VSClinical, the process of scoring the ACMG guidelines requires evaluating evidence for the connection between a variant and the disorder or condition being evaluated by the genetic test for an individual. These lines of evidence cover clinical presentation, gene function, bioinformatic annotations and in-silico… Read more »
In the last installment of this series, I covered the basics of variant interpretation and how it fits into the genomic testing process. Now we can cover in more detail how VSClinical works, what algorithms and annotation sources power the recommendations and how the ACMG criteria are organized into useful categories. VSClinical is built to make the process of evaluating… Read more »
This week we launched VSClincial with our first webcast to show our powerful new way to perform variant interpretation following the ACMG guidelines. Our audience asked a lot of great questions on the new product and I’d like to highlight a few here. Can VSClinical run on a laptop and/or a locked down environment? Like all of Golden Helix products, you have… Read more »
Yesterday we launched VSClincial with our first webcast in what will be a series about this powerful new way to perform variant interpretation following the ACMG guidelines. In this post, I wanted to cover the motivation for VSClinical and how we curated and presented the 33 criteria from the ACMG Guidelines into an intuitive workflow with various bioinformatic evidence and… Read more »
There are many good reasons why the pursuit of the highest quality genomic interpretation would lead you to the latest human reference. It is more complete and fixes incorrect or partially missing genes that have known implications for human disease. While most major projects cataloging human populations have plans to re-do all their genomic alignments to the new human reference… Read more »
The Golden Helix SNP & Variation Suite (SVS) platform is a powerful and versatile set of tools and algorithms for performing genomic research. That research spans from data originating on genotype micro-arrays to next-generation sequencing. While the majority of SVS users start with genotype data on their samples, any genomic information across a cohort can be used in our various… Read more »
We have a lot to thank the 1000 Genomes project for in the genomics community. By the collaborate efforts of many researchers and organizations, the project produced not only the first catalog of rare human variation but in the process standardized many things we take for granted, such as the VCF and BAM file formats. The variant frequencies of the… Read more »
With the recent release of VarSeq 1.4.7, we have expanded the concepts of our popular assessment catalog to include CNV and other region-based records and not just variants. To match these capabilities, we have made a major update to VSWarehouse that supports these new record types in the centrally hosted and versioned Catalogs and Reports. Review of the VSWarehouse Genomic… Read more »
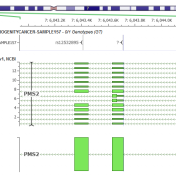
VarSeq Updated with CNV Annotations, CNV PhoRank, and Region Assessment Catalogs This year we have released multiple advances to support CNV analysis in VarSeq, expanding our target region based VS-CNV caller to handle exomes and low-depth genomes as well as additional supporting algorithms like calling Loss of Heterozygosity regions. To top this off, VarSeq 1.4.7 has been shipped with many… Read more »
The VarSeq clinical platform is built on a strong foundation of data curation and annotation algorithms to ensure the variants identified have all the information required to make the correct clinical assessments. It’s easy to make light of “variant annotation”, but the details run very deep into the roots of how we represent genomic data, how public data is aggregated, stored… Read more »
When studying complex diseases that may have multi-genic contributions from across the genome, it is not uncommon to see what may appear like elevated correlation between your trait or other test variable and the SNPs across the genome. The problem is at first glance you won’t be able to tell if this is due to a population structure in your… Read more »
We have been heads down doing the detailed and careful work to improve our CNV caller algorithm in the past three months since our we launched our Exome capable CNV caller and are very excited about the massive step forward we have made with the VarSeq 1.4.5 release. Additionally, we have added the all new Whole Genome large-event caller capable… Read more »
Annotating with gnomAD: Frequencies from 123,136 Exomes and 15,496 Genomes When the Broad Institute team lead by Dan MacArthur announced at ASHG 2016 that the successor to the popular ExAC project (frequencies of 61,486 exomes) was live at http://gnomad.broadinstitute.org/, I thought their servers would have a melt-down as everyone immediately jumped on and started looking up their favorite genes and… Read more »
Recently, we added a natively supported Genotype Phasing and Imputation capability in SNP & Variation Suite 8.7.0. Since then we have had fantastic feedback and adoption as folks take advantage of the BEAGLE 4.0 and 4.1 algorithms from within their existing SNP GWAS and agrigenomic workflows. One piece of feedback we heard from our time at PAG, ACMG and our… Read more »
It may be possible to say that annotating a variant correctly and accurately against gene transcripts is the most important job of a variant annotation and interpretation tool. We take it very seriously at Golden Helix as we support VarSeq and its use by our customers in both research and clinical contexts. It has been a source of frustration that… Read more »
In the past couple of weeks, the topic of the Filter and Quality fields in the popular ExAC population catalog has come up a number of times. It turns out that unlike the 1000 Genomes project, which decided to very heavily filter their variant list to only contain variants they consider high quality, ExAC chose to include more dubious variants… Read more »