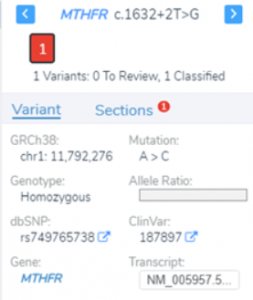
Transitioning to Broad-Scale Genomics: From Gene Panels to Whole Genome Sequencing
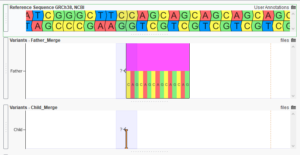
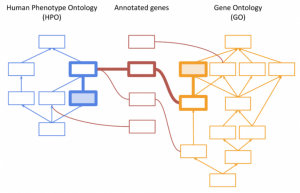
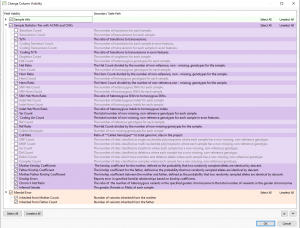
Explore the evolving landscape of genomic analysis, transitioning from targeted gene panels to whole genome sequencing. A recent trend with our customers has been to expand their workflows from small panel sequencing analyses to larger whole exome and genome sequencing analyses. The decreasing cost of sequencing has made this a…
Read more →