Advanced Techniques for GenomeBrowse Customization

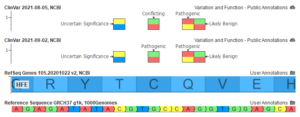
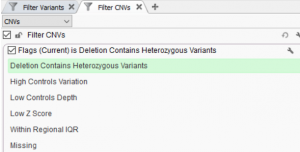
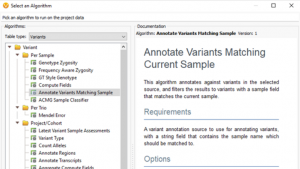
Today, we will be exploring the indispensable role of GenomeBrowse in your VarSeq workflows. This blog aims to guide you through various customization options, including color preferences, filtering techniques, display modifications, numeric value plotting, and additional tips. I hope to enhance your GenomeBrowse experience and enable you to gain valuable…
Read more →