New discoveries using NGS data analysis are never-ending and are pushing precision medicine to the forefront. In this month’s customer publication blog, I am focusing on our VarSeq software as investigators harness its power to perform a variety of investigational study designs. From cancer to inherited rare disease research, VarSeq is the rising star in research and diagnostic tools. At… Read more »
I am pleased to share with you the official release of my updated eBook, “Precision Medicine“. Almost 2,500 years ago, Hippocrates captured one of the key principles underlying precision medicine. In the 21st century, we take an understanding of the individual characteristics of a person to a new level. By leveraging information about an individual’s genome, we are able to… Read more »
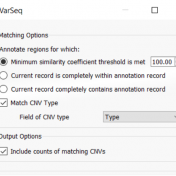
In continuation of our blog posts focusing on new features of VarSeq v2.2.2, here we will discuss the Latest Sample Assessment algorithm for both single nucleotide variants (SNVs) and copy number variants (CNVS). This algorithm annotates the variants of the project with the latest assessment from your variant catalog, which will show the history of interpretations made for the variants… Read more »
Golden Helix offers a market-leading bioinformatics solution that allows users to evaluate next-generation sequencing variants according to the ACMG and now ACGS guidelines. The ACMG guidelines were created in 2015 and are widely accepted as best practice for the interpretation of sequencing variants throughout the United States (Richard et al 2015). Very similar are the ACGS guidelines that were developed… Read more »
VarSeq 2.2.2 was released on December 17th, 2020 and the main feature that was added to VarSeq was that the VSClinical ACMG Guidelines workflow now has an additional CNV interpretation framework based on the ACMG/ClinGen guidelines. This product supports interpreting CNVs detected with VS-CNV or imported CNVs alongside variants and requires both a VSClinical ACMG license and a CNV license…. Read more »
Reading scientific articles that our customers have recently published is one of my favorite things here at Golden Helix. It is fascinating to learn about the research and to see the various ways our software gets put to the test.Since we are rolling out our most extensive VarSeq update yet, I thought it would be a great time to look… Read more »
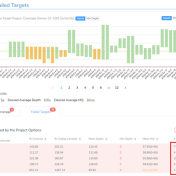
Curious about how coverage statistics can be used in conjunction with VarSeq? Evaluating the coverage over target regions or whole genomes is essential whether you are working with variant or CNV analysis. VarSeq has had the capability to compute sample level coverage statistics for some time now, but in the 2.2.2 release of VarSeq, there are some new features that… Read more »
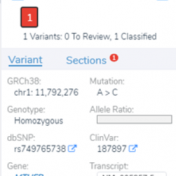
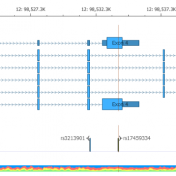
In this blog post, I will be analyzing a loss-of-function splice variant in MTHFR using VarSeq. In the search for clinically relevant variants contributing to rare disorders, efficient filtering strategies are an important step in eliminating disinteresting variants. However, any applied filters must also ensure no interesting variants inadvertently get filtered out. Golden Helix provides the tools to complete this… Read more »
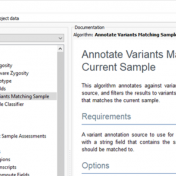
Those of you who have been attending our recent webcasts have learned about our upcoming VarSeq release. A part of that release will be an additional algorithm that will annotate variants matching the current sample. If you are not familiar with these webcasts, here are several on-demand webcasts I recommend to get you familiar with these new features: Evaluating Copy… Read more »
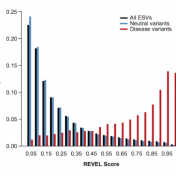
Typically, researchers are looking for rare variants in their next generation sequencing datasets. However, most of the nonsynonymous variants have unknown significance because there is an inherent difficulty in validating large numbers of rare variants or even detecting rare variants with high statistical power. In lieu of this issue, computational tools are needed as they accurately predict the pathogenicity of… Read more »
A common discussion with our customers includes the challenges with the tertiary stage of analyzing next-gen sequencing data. This is the stage where all data from gene panels, exome, or whole genome scale pass through filters to quickly isolate the clinically relevant variant contributing to a patient disorder. Golden Helix has recognized these challenges in the scale of data and… Read more »
In the 1990s the genetic industry voiced a request for a variant catalog that incorporates associated variant information such as phenotypic and metabolic pathways. The call was answered by NCBI, which created dbSNP; dbSNP became publicly available in 1998 and around 1.5 million variants. Fast forward to the present and dbSNP now contains over 2 billion SNPs spanning human, rat,… Read more »
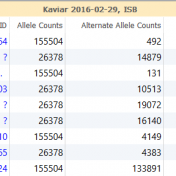
In the search for disease causing mutations it is important to determine if the variant has been previously observed in humans and at what frequency. With the advent of increasing genomic information, there is now a variety of different databases and annotation sources that can be utilized. For some, this could be a tedious task that leads only to implementing… Read more »
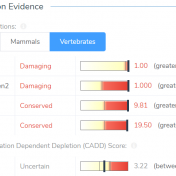
The University of Washington’s Combined Annotation Dependent Depletion (CADD) algorithm measures the deleteriousness of genetic variants. This includes single nucleotide polymorphisms (SNVs) and short insertions and deletions (indels) throughout the human reference genome assembly. This algorithm was introduced in 2014 and has since become one of the most widely used tools to assess human genetic variation. Since 2014, the algorithm has been… Read more »
An under-appreciated area of complexity when looking into the field of genetics from the outside can be found in genes and transcripts. Alternative splicing allows eukaryotic species to have a wonderfully powerful genetic code, resulting in multiple protein isoforms being encoded in a single section of DNA. But when it comes to variant interpretation, different transcripts can result in widely different predicted… Read more »
In VarSeq 2.2.1, you can set template annotation sources to automatically update to the latest version. Previously, VarSeq templates were frozen in time. Now, each new project created from a template would use the same source that was used when the template was created. When you save a template, you can have the sources automatically update to the latest version…. Read more »
Thank you to everyone who joined me for our latest webcast, “Next-Gen Sequencing of the SARS-CoV-2 Virus with Golden Helix.” If you missed the live event and are interested in knowing what we talked about, you may access the recorded event below: Our Live Q&A generated a lot of great questions. Unfortunately, we were unable to answer them all, but… Read more »
VarSeq 2.2.1 was released on April 1st and features an upgraded gene annotation capability with new RefSeq genes tracks and an AMP workflow addition: the Drugs and Trials tab. The new RefSeq human genome genes tracks contain updated gene names and the recognition of any MANE (Matched Annotation from NCBI and EMBL-EBI) identified transcripts. VarSeq has been updated to be… Read more »
Generating a clinical report is the final step of most NGS pipelines and is important as it relays results and information to legacy systems, physicians and ultimately the patient. As reporting is a valuable process, Golden Helix offers reporting capabilities according to the ACMG and AMP guidelines but also as a standalone feature in VSReports. VSReports is a platform that… Read more »
At Golden Helix, we want our new users to hit the ground running with VarSeq and not spend oodles of time getting started building and automating their workflows. To achieve this goal, our team has generated blogs, webcasts, and tutorials that explain and demonstrate workflows that are possible with VarSeq. Each VarSeq tutorial offers step by step instruction in which… Read more »