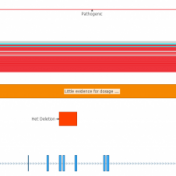
With the latest release of VarSeq, we have made significant updates to our handling of the interaction of variants and genes. This includes the support for non-coding transcripts, improved splice site predictions, and updates to gene and transcript annotations. We received several questions regarding how decisions are made in the software regarding genes and transcripts with these gene-related changes. This… Read more »
While VarSeq has always had excellent support for variant interpretation and analysis, we continue to find new edge cases in the clinical literature that improve our interpretation capabilities. In this blog, we will be covering some of the new improvements in VarSeq to support the interpretation of non-coding and splice site variants. Transcript Annotation Improvements Let’s start by covering some… Read more »
Thank you to those who attended the recent webcast, “Exome Analysis with VS-CNV & VSClinical: Updated Strategies & Expanded Capabilities”. For those who could not attend but wish to watch, here is a link to the recording. In this webcast, we covered the capabilities and updates that have been incorporated into VarSeq that enhance whole exome sequencing workflows. The new… Read more »
In this blog, we will be covering new assessment catalogs and how they work to improve saving and tracking variant interpretations. VarSeq is a variant analysis tool that effectively analyzes single nucleotide (SNVs) and copy number variants (CNVs) in both cancer and germline workflows. Because VarSeq enables such diverse variant analysis, there are many research labs and institutions that evaluate… Read more »
Thank you to those who attended the recent webcast, “VSWarehouse: Tracking Changing Variant Evidence and Classifications”. For those who could not attend but wish to watch, here is a link to the recording. The webcast covered some general highlights of VSWarehouse value but also presented some specific capabilities covering the ClinVar classification tracker. Golden Helix provides complete solutions to handle… Read more »
The recent release of VarSeq 2.2.2 brings our Word report template system, previously featured in VSClinical AMP, to the VSClinical ACMG workflow. This blog post will describe how to use the Word template system using one of our shipped templates as well as how to start customizing your own templates. We will cover the three different report templates that ship… Read more »
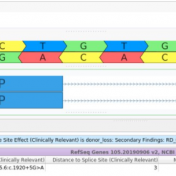
Our latest VarSeq release is one of the largest we’ve ever had, boasting an extensive list of new features and improvements. As part of this release, we have dramatically expanded our support for splice site analysis. This includes improvements to our novel splice site algorithm and support for splice site effect prediction along with several other small improvements. Novel Splice… Read more »
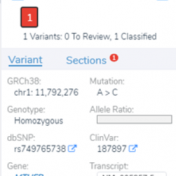
In continuation of our blog posts focusing on new features of VarSeq v2.2.2, here we will discuss the Latest Sample Assessment algorithm for both single nucleotide variants (SNVs) and copy number variants (CNVS). This algorithm annotates the variants of the project with the latest assessment from your variant catalog, which will show the history of interpretations made for the variants… Read more »
Didn’t catch the webcast live? No worries! We cover ‘VSClinical: A Complete Clinical Workflow Solution’ Q&A’s in this blog post. The webcast, ‘VSClinical: A Complete Clinical Workflow Solution’ demonstrated how solutions provided by Golden Helix can be implemented to cover all requirements of a clinical workspace. Specifically, this webcast focused on a detailed workflow from a bioinformatician, geneticist, and lab… Read more »
Reading scientific articles that our customers have recently published is one of my favorite things here at Golden Helix. It is fascinating to learn about the research and to see the various ways our software gets put to the test.Since we are rolling out our most extensive VarSeq update yet, I thought it would be a great time to look… Read more »
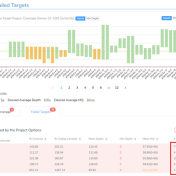
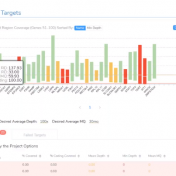
Curious about how coverage statistics can be used in conjunction with VarSeq? Evaluating the coverage over target regions or whole genomes is essential whether you are working with variant or CNV analysis. VarSeq has had the capability to compute sample level coverage statistics for some time now, but in the 2.2.2 release of VarSeq, there are some new features that… Read more »
Webcast Recap In the recent webcast “Exploring New Features and Clinical Reports in the ACMG Guideline Workflow”, Gabe and I took viewers through an evaluation with CNVs and SNVs according to the ACMG Guidelines where we generated and customized a clinical report. Along the way, we highlighted many new features that will soon be available in the upcoming VarSeq release…. Read more »
In this blog post, I will be analyzing a loss-of-function splice variant in MTHFR using VarSeq. In the search for clinically relevant variants contributing to rare disorders, efficient filtering strategies are an important step in eliminating disinteresting variants. However, any applied filters must also ensure no interesting variants inadvertently get filtered out. Golden Helix provides the tools to complete this… Read more »
In our previous webcast, Evaluating CNVs with VSClinical’s New ACMG Guidelines, we focused on a CNV deletion (12:27715515-29628122×1) in which the patient had a known disorder called Brachydactyly type E. The CNV was isolated using our VS-CNV caller and applied to the ACMG CNV guidelines using the intuitive steps of VSClinical. If you missed the webcast, you can watch the… Read more »
In a recent webcast, users were exposed to some new features upcoming in the next release of VarSeq. In this update, we will use an example de Novo CNV in a cardiomyopathy panel in VarSeq. There is a long list of new tools and polishes to the software but one major upgrade is the inclusion of the ACMG and ClinGen… Read more »
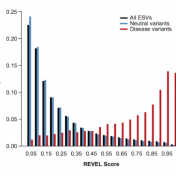
Typically, researchers are looking for rare variants in their next-generation sequencing datasets. However, most of the nonsynonymous variants have unknown significance because there is an inherent difficulty in validating large numbers of rare variants or even detecting rare variants with high statistical power. In lieu of this issue, computational tools are needed as they accurately predict the pathogenicity of rare… Read more »
A common discussion with our customers includes the challenges with the tertiary stage of analyzing next-gen sequencing data. This is the stage where all data from gene panels, exome, or whole genome scale pass through filters to quickly isolate the clinically relevant variant contributing to a patient disorder. Golden Helix has recognized these challenges in the scale of data and… Read more »
In this blog update, I’ll be walking you through some of the advanced plotting capabilities with GenomeBrowse. The strategy with any next-gen sequencing analysis is to filter down to interesting variants for either research or clinical conclusion. Golden Helix produces powerful software specifically tailored for this efficient and comprehensive search for interesting and clinically relevant variants. One additional advantage of… Read more »
In our recent webcast announcing the upcoming release of VarSeq VSClinical and the implementation of the ACMG guidelines for NGS CNVs, we had a number of live questions we didn’t get a chance to cover at the end of the presentation. I will follow up on those questions in this blog post. But first, if you didn’t get a chance to join us for… Read more »
In the 1990s the genetic industry voiced a request for a variant catalog that incorporates associated variant information such as phenotypic and metabolic pathways. The call was answered by NCBI, which created dbSNP; dbSNP became publicly available in 1998 and around 1.5 million variants. Fast forward to the present and dbSNP now contains over 2 billion SNPs spanning human, rat,… Read more »