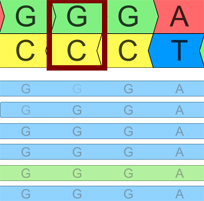
FAQ: Creating Repeatable Clinical Workflows
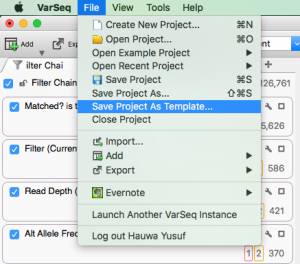
Question: Now that I’ve added annotation sources for my sample, filtered down to a list of interesting variants, flagged those variants and generated a clinical report, can I save or copy the annotation sources and filters for use on another sample? Short Answer: Yes! Long Answer: VarSeq was created with…
Read more →