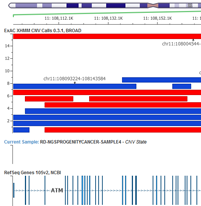
Calling Large LOH and CNV Events with NGS Exomes

We are pleased to announce our next webcast, Calling Large LOH and CNV Events with NGS Exomes. The live event is scheduled for Wednesday, March 8th at noon EST. Here are the specifics: Wednesday, March 8th 12:00 pm EST Next Generation Sequencing Exomes are a powerful assay used in both clinical…
Read more →