Loss-of-Function Splice Variant in MTHFR
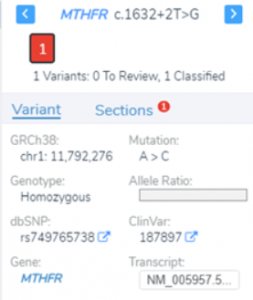
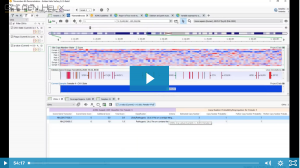
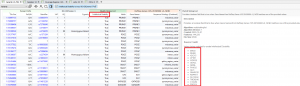
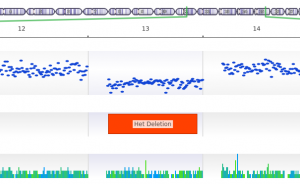
In this blog post, I will be analyzing a loss-of-function splice variant in MTHFR using VarSeq. In the search for clinically relevant variants contributing to rare disorders, efficient filtering strategies are an important step in eliminating disinteresting variants. VarSeq supports end-to-end rare disease analysis, from panel-level filtering to clinical variant…
Read more →